molekularna biologija
Googleova umjetna inteligencija riješila dosad nerješive probleme u molekularnoj biologiji.
 Tamara Čačev / 2. prosinca 2020. / Perspektive / čita se 13 minuta
Tamara Čačev / 2. prosinca 2020. / Perspektive / čita se 13 minuta

Tamara Čačev / 2. prosinca 2020. / Perspektive / čita se 13 minuta
Koliko se radi o superiornom načinu rješavanja problema govori to što je AlphaFold za samo pola sata riješio strukturu bakterijskog proteina koja se eksperimentalno rješavala gotovo desetljeće, objašnjava Tamara Čačev. Taj je algoritam sudjelovao i u predikciji strukture proteina SARS-Cov-2 virusa i kada je za nekoliko mjeseci dobivena struktura nekih od njih predviđanje je bilo prilično vjerodostojno
Ovih dana medije je poharala vijest o još jednoj pobjedi „umjetne“ inteligencije, Deep Mind algoritam AlphaFold, projekt Google AI napravio je možda mali korak za njega, ali velik za čovječanstvo i počeo rješavati do sada gotovo nerješive probleme u molekularnoj biologiji. Mislite da je hodanje po Mjesecu bitno, e pa nije barem u onom doslovnom smislu ako izuzmemo enormni napredak tehnologije koji je takav projekt donio sa sobom. Ovo je stvarni iskorak koji ćemo i vi i ja osjetiti na svojoj koži već za naših života. Umjetna inteligencija je odrasla, ne igra više samo društvene igre tipa Go, već rješava stvarne probleme – uspješno je predvidjela trodimenzionalnu strukturu proteina na temelju linearnog niza aminokiselina u proteinskom lancu.1 Vauu! Priznajte ništa vam nije jasno, koga bi to uopće trebalo zanimati. Stoga krenimo od početka.
Kada se razmišlja o molekularnoj biologiji i genetici prve asocijacije su geni i molekule DNA. I naravno sve se vrti oko tog magičnog zapisa u kojem se nalazi uputstvo za život svega što jest živo na Zemlji, pa čak i virusa koji „ožive“ unutar naših stanica zahvaljujući vlastitom receptu za „život“ koji je također zapisan u njihovom genomu. No, kad se odmaknemo od škole, puno toga ispari pa tako i to da molekula DNA koliko god čudesna bila nije sama sebi svrhom nego je kod za „nešto“. To „nešto“, većini ljudi se pogubi putem a jednako je bitno. Ultimativni produkt zapisa u genomu nekog organizma su molekule RNA i proteini. Nekad se mislilo da ima svega nekoliko tipova molekula RNA, danas znamo da nije tako, no o tome drugom prilikom. Ipak ono što znamo jest da su sve RNA u konačnici direktno ili indirektno uključene u sintezu proteina i/ili njihovu pravilnu vremensku i prostornu regulaciju.
Kad kažemo proteini, većini ljudi je to asocijacija na piramidu zdrave prehrane kao jedan od tri stupa makronutrijenata koji su nam neophodni za život. Znate ono, meso, mlijeko, jaja i grahorice. Naravno, u našem probavnom sustavu oni se razlažu na osnovne građevne jedinice aminokiseline koje smo u stanju „upiti“ u krvotok i raspodijeliti po organizmu za izgradnju vlastitih proteina, naše kože, kose, noktiju, mišića itd. No osim ove građevne funkcije koja nam je svima očita i razumljiva proteini u našem organizmu rade sve – oni su glavni egzekutori većine naših funkcija od razine stanice do cijelog organizma. Poremećaji u njihovoj funkciji od našeg rođenja uzrok su mnogih nasljednih bolesti te ako se radi o nekoj esencijalnoj životnoj funkciji tada mutacije u genu koji kodira za protein mogu biti nespojive sa životom. Kasnije, tijekom života opet dolazi do akumulacije grešaka u kodu za regulaciju sinteze proteina, u zapisu samog proteina ili u mehanizmima za njegovo uklanjanje kad je izvršio funkciju ili je oštećen. Kad razmišljamo što je to bolest na molekularnoj razini gotovo svim bolestima uzrok je disfunkcionalni protein ili njegova neadekvatna količina na krivom mjestu. Jedan od koncepata kako se zapravo objašnjava starenje jest akumulacija oštećenja genetskog materijala koja rezultira neadekvatnom funkcijom proteina ili njihovim neadekvatnim uklanjanjem kad su oštećeni. Taj proces akumulacije mutacija i oštećenja proteina najbolje se vidi ako usporedimo kožu lica novorođenčeta i čovjeka u poznim godinama. Sve je to oštećenje proteina od UV zračenja, pušenja, alkohola, vanjskih uvjeta, prehrane, ukratko života. Ovo je samo estetika, no takvi se procesi odvijaju i u nutrini organizma što je za naše preživljavanje puno bitnije. Tumorske bolesti su notorni primjeri disfunkcije proteina koji se zbog mutacija nepravilno sintetiziraju ili u krivo vrijeme ili na krivom mjestu. Dijabetes je rezultat disfunkcije inzulina ili njegovih receptora koji su također proteini. Da zaključimo, u molekularnoj osnovi većine bolesti jest disfunkcija proteina.
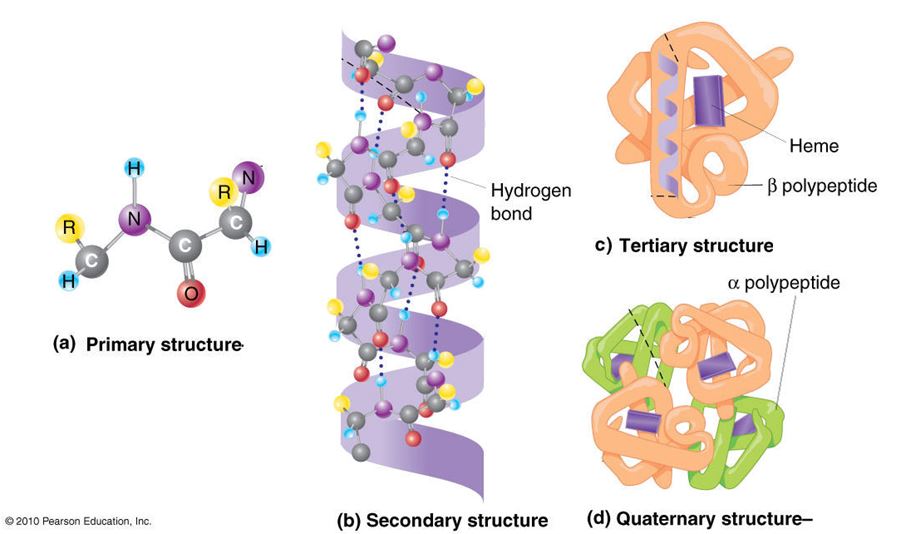
Ovo prije navedeno su razlozi zašto je bitno proučavati proteine na njihovoj elementarnoj razini i zašto su neki ljudi posvetili cijele svoje znanstvene karijere baveći se možda jednim proteinom cijeli život. No, vratimo se natrag na bazičnu znanost. Na slici 1. prikazan je tipičan protein, u kemijskom smislu radi se o lancu međusobno povezanih aminokiselina koje su se točno određenim redom nanizale prema uputi koja piše u genu za taj protein. Na najelementarnijoj razini radi se o linearnom slijedu koji se, zahvaljujući tome što je dešifriran genetski kod, može relativno jednostavno pročitati već iz same molekule DNA. Naravno, možemo ići i obrnuto, izolirati neki protein i identificirati ga no taj pristup je neusporedivo teži, skuplji i dugotrajniji a često i s upitnim ishodom zbog metodologije koja je puno kompleksnija u proteinskoj kemiji. Odlično, imamo proteinsku sekvencu, što nam ona može reći o funkciji proteina? Danas više nego u počecima molekularne biologije, ali zapravo u slučaju manje poznatih proteina sa specifičnim funkcijama u organizmu i dalje tapkamo u mraku. Treba imati na umu da i danas za veliki broj proteina u našem organizmu koje smo identificirali nemamo pojma što zapravo rade. Ono što se danas radi zahvaljujući prodoru bioinformatike u analize genoma jest da se određena sekvenca koja kodira za neki protein pokuša pronaći kroz usporedbe sa sekvencama koje su dešifrirane u drugim organizmima. Ukoliko se radi o nekom proteinu koji je odgovoran za neke najelementarnije funkcije na razini stanice tada je velika vjerojatnost da će njegova sekvenca zbog svoje važnosti biti prisutna i u najprimitivnijim oblicima života, poput bakterija, i da će biti vrlo evolucijski očuvana s velikom sličnošću našoj verziji proteina. S druge strane, kod za prije spomenuti inzulin nećemo naći u bakterijama jer je funkcija koju on vrši potrebna višim organizmima kod kojih postoji hormonalna regulacija i koordiniranje rada stanica.
No, proteini u organizmu ne dolaze kao linearne molekule već njihova funkcija prolazi iz njihove trodimenzionalne strukture. Prisjetimo se paradigme ključa i ključanice koja se često koristi u udžbenicima kako bi se objasnilo kako enzimi (opet se radi o proteinima) „prepoznaju“ molekule na koje moraju djelovati kao i mjesto na kojem će djelovati. U toj paradigmi samo molekule koje svojom trodimenzionalnom strukturom (ključ) mogu ući u ključanicu koja je metafora za trodimenzionalno mjesto koje svojom strukturom napravi enzim, mogu biti dalje procesirane. Ovaj princip vrijedi i za druge protein-protein interakcije, jedna od danas iznimno aktualnih je i kako se SARS-Cov-2 svojim proteinom Spike veže na receptor (protein) na našim stanicama te zbog toga što pogađa točno njegovu strukturu „otključava“ svoj ulaz u stanicu.
Biološka funkcija proteina proizlazi dakle iz njegove trodimenzionalne strukture. Nažalost, izolirati protein u njegovoj nativnoj trodimenzionalnoj strukturi iznimno je težak, mukotrpan i često jalov posao jer postupci koji se pri tome koriste uništavaju tu strukturu. Ipak metodama kristalografije uspješno je razotkrivena trodimenzionalna struktura nekih bitnih proteina u stanici. Na temelju tih pionirskih istraživanja proteina koja se protežu unatrag gotovo čitavo stoljeće imamo neke ideju o tome kako se proteini „slažu“ u prostoru. Nakon primarne strukture koja je linearni slijed aminokiselina, dijelovi proteinskog lanca zauzimaju određeni 3D oblik te se smotavaju u elemente sekundarne strukture koje nalikuju na zavojnice ili ploče. Zatim se ti elementi sekundarne strukture dalje slažu u prostoru u elemente tercijarne strukture. Kada se protein sastoji od više podjedinica odnosno lanaca tada se elementi tercijarne strukture u prostoru međusobno slažu tako da zauzmu kvartarnu strukturu koja je na kraju jedina funkcionalna (slika 1).
Jesu li nam poznata pravila po kojima se odvija ovo prostorno slaganje u tri dimenzije. Djelomično jesu jer je sigurno da su rezultat onoga što je zapisano na linearnoj razini. Struktura u prostoru mora proizlaziti iz linearnog slijeda aminokiselina u proteinskom lancu. To je postulat kojeg je prilikom primanja Nobelove nagrade za kemiju 1972. godine izrekao i Christian Anfinsen. Sile i veze koje se javljaju kada se u prostoru susretnu dijelovi lanca koji u sekvenci nisu blizu kemijske su ili elektrostatske prirode. No, možemo li to prepoznati, pročitati i predvidjeti iz linearnog slijeda? Što to zapravo znači? Možemo li predvidjeti kako će se u tri dimenzije smotati linearni proteinski lanac. Zamislite koliko postoji mogućnosti da se na nekoj vezici poredane kuglice poslože u prostoru u neku 3D strukturu. U redu, nije to bez nekog reda, poštuju se neke kemijske, elektrostatske i u konačnici energetske zakonitosti ali čak i tada mogućnosti su bezbrojne i to je osnova problema koji je u međuvremenu dobio i svoje ime „protein folding problem“ i zagorčao život mnogim znanstvenicima koji su na njemu tijekom godina radili. Mogućnosti je naime toliko (10300 kombinacija) da bi trebalo vrijeme od postanka svemira do danas da se računanjem identificiraju sve moguće konfiguracije tipičnog proteina, a ipak u stanici se on posloži u pravilnu strukturu u nekoliko milisekundi. Ovaj se fenomen naziva Levinthalovim paradoksom po Cyrusu Levinthalu koji ga je 1969. i opisao. Stoga je jasno da je predikcija proteinske strukture iz sekvence predstavljala većinom Sizifovski desetljeća dug posao i gotovo nerješiv problem za ogroman broj proteina koji postoji u živim organizmima unatoč napretku u različitim modeliranjima ovih procesa.
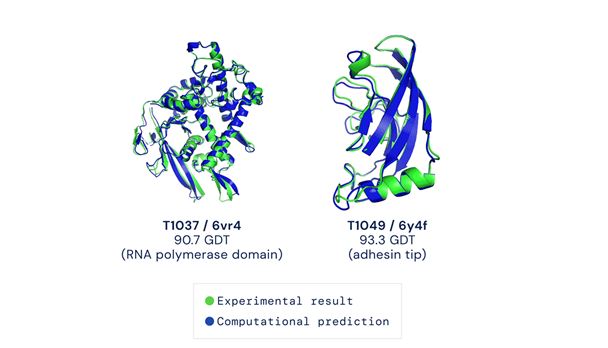
Sve dok se nije pojavio DeepMind AlphaFold algoritam koji to radi čini se jako uspješno. Zamislite sad nekog umirovljenog kristalografa koji je radni vijek potrošio na možda nekoliko proteinskih struktura, je li sretan ili mu je mučno. DeepMindov algoritam AlphaFold2 nadmašio je u predikciji proteinskih struktura stotinu drugih timova koji su se natjecali na izazovu „Critical Assessment of Structure prediction“. Radi se o natjecanju koje se održava od 1994. godine s ciljem poboljšavanja metoda računalne biologije i što boljeg predviđanja proteinskih struktura. Skupine dobivaju zadatak predvidjeti strukture proteina koje su dobivene eksperimentalno ali većinom još nisu objavljene. Time se postiže da sustav bude „naivan“ za bilokakve „sugestije“ na temelju poznatih činjenica. U ovom natjecanju AlphaFold je bez greške predvidio neke od eksperimentalno dobivenih struktura metodom kristalografije uz pomoć redgenskih zraka i krio-elektronske mikroskopije. Prije dvije godine prva verzija ovog algoritma također je sudjelovala na ovom izazovu i tada je umjetna inteligencija odnosno deep learning korištenjem strukturalnih i genetičkih podataka predviđao udaljenost između pojedinih aminokiselinskih parova u proteinu u 3D. U ovogodišnjoj verziji koriste se podaci o fizikalnim i geometrijskim ograničenjima u prostornoj organizaciji proteinskog lanca i predviđa se konsenzusni model čitavog proteina a ne međuodnos pojedinih aminokiselina. To je veliki iskorak prema upotrebljivosti ovih predviđanja. Na ovogodišnjem izazovu trebalo je riješiti stotinjak struktura i neke je AlphaFold2 predvidio bolje a neke lošije no gotovo dvije trećine dobivenih predikcija bile su po kvaliteti usporedive onim dobivenim eksperimentalno (slika 2). O detaljima samog procesa stvaranja modela „prostornih grafova“ koji je dobiven kroz iteracijske procese učenja 170 000 proteinskih struktura više je podataka u referenci 2. Ono gdje je ove godine podbacio bile su strukture koje su dobivene nuklearnom magnetskom rezonancijom, trećom metodom koja se koristi u eksperimentalnoj analizi 3D strukture proteina pretpostavlja se zbog načina kako se sirovi podaci dobiveni ovom metodom „pretvaraju“ u model.
AlphaFold 2 nije bio jedini deep learning algoritam koji je korišten na ovom izazovu, zapravo je polovina natjecatelja koristila neki od svojih deep learning algoritama, no AlphaFold 2 je bio najuspješniji. Koliko se radi o superiornom načinu rješavanja problema u odnosu na dosadašnje mogućnosti govori primjer kako je AlphaFold za svega pola sata riješio strukturu bakterijskog proteina koja se eksperimentalno rješavala gotovo desetljeće. Ovaj je algoritam sudjelovao i u predikciji strukture proteina SARS-Cov-2 virusa koji nisu bili prethodno eksperimentalno analizirani, i s pomakom od nekoliko mjeseci kada se dobila struktura nekih od njih predviđanje je bilo prilično vjerodostojno.2 To ne znači da će eksperimentalci u proteinskoj kemiji u mirovinu, no dobit će ekstremno moćno oruđe za ubrzavanje i usmjeravanje svojih istraživanja. Jer kao što smo rekli, još je jedno veliko more, zapravo ocean proteina u našem genomu o kojima ne znamo ništa osim da postoje. Danas na razini zapisa imamo identificirano oko 180 milijuna proteinskih sekvenci no u strukturalnom smislu riješeno je zasad 170 000 proteina. Također, neke proteine poput membranskih koji imaju važnu ulogu u komunikaciji stanica iznimno je teško/gotovo i nemoguće kristalizirati zbog njihovih intrinzičkih svojstava. U ovom moru još nerješenih proteina svi su sigurno bitni, no možda su neki „bitniji“ za rješavanje najvećih zdravstvenih problema današnjice. Umjetna inteligencija enormno će ubrzati ovu potragu.
Objasnili smo da je bitno znati trodimenzionalnu strukturu proteina jer je ona ključna za dešifriranje njegove uloge u organizmu, odnosno kako neki protein radi to što radi. To jest i nije fundamentalno pitanje jer iako se tako ne čini na prvi pogled, odgovor može imati direktnu primjenjivost ponajprije u razvoju ciljanih lijekova za niz bolesti gdje je ključni element disfunkcija proteina, dakle gotovo u svima. Kada znamo trodimenzionalnu strukturu proteina, kako u prostoru izgleda neki njegov dio bitan za funkciju, možemo prema njemu modelirati molekulu lijeka da precizno uđe u to aktivno mjesto i da ga npr. inhibira. To naravno nećemo raditi s proteinima koji su funkcionalni, ali možda bismo mogli ciljano blokirati receptore na tumorskim stanicama što se uostalom već i radi na temelju empirije ali sve više i korištenjem dosadašnjih modela predikcije. Uvođenjem umjetne inteligencije, problem trodimenzionalne strukture mogao bi se rješavati brže i bolje što posljedično vodi u čitav spektar novih meta na koje se može djelovati ciljanim lijekovima. Sa druge strane računalna kemija i predikcije različitih malih molekula koje će najbolje odraditi takav posao predstavljaj medalje ovog procesa napadaju isti problem s druge strane čime se proces enormno ubrzava jer se dio rješenja eliminira već in silico. Sličnim ubrzanim tijekom događanja kroz bioinformatičku analizu sekvence genoma virusa SARS-Cov-2 (što nije ni približno toliko kompleksan problem kao rješavanje 3D sturkture proteina) došlo se u svega nekoliko tjedana do mete za razvoj cjepiva koja je pokazala učinkovitost gotovo bez presedana. Ne treba ipak imati iluzije da su sada svi problemi nastanka novih terapija riješeni jer od modeliranja in silico do potvrđenog registriranog lijeka i dalje je velik put testiranja in vitro i in vivo, no barem smo ovaj prvi dio pronalaska prave mete i potencialno prave molekule za terapiju možda ipak skrati i/ili pametnije usmjeri u odnosu na puki princip pokušaja i pogreške.
In Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstract Book), 30 November – 4 December 2020. https://predictioncenter.org/casp14/doc/CASP14_Abstracts.pdf
 5.00
(4)
5.00
(4)
7. travnja 2026. / U fokusu

3. travnja 2026. / U fokusu

31. ožujka 2026. / U fokusu

25. ožujka 2026. / U fokusu

7. travnja 2026. / Perspektive Publikacije

2. travnja 2026. / Perspektive Publikacije

2. travnja 2026. / Perspektive Publikacije

1. travnja 2026. / Perspektive Publikacije
