-
-
dr. sc. Tamara Čačev viša je znanstvena suradnica u Zavodu za molekularnu medicinu Instituta Ruđer Bošković. Čačev je također članica Savjeta Ideje.hr
Kada sam početkom prošle godine napisala prvi tekst o SARS-CoV-2 virusu i COVID-19 vjerojatno nisam bila do kraja svjesna razmjera koje će epidemija poprimiti i kakve će duboke tragove ostaviti na svakome od nas. Danas, godinu dana nakon prvih službeno evidentiranih slučajeva ove bolesti, protekla godina čini se kao kontinuirana noćna mora koja je osim posljedica na zdravlje, aktivirala i naše najesencijalnije egzistencijane strahove koji su u doba blagostanja nekako potisnuti na margine naše svijesti. Ipak, ima i nešto dobro u protekloj godini, pokazalo se kako je molekularna biomedicina u vrlo kratkom vremenu suvereno dala rješenja za svaki problem koji se pojavio tijekom epidemije, od izolacije virusa i njegovog sekvenciranja, molekularnih metoda njegove detekcije koje su postale mainstream i u najvećoj zabiti na ovom planetu, što je presedan sam po sebi, sve do brzog pronalaska mete za razvoj pametnog cjepiva i to na nekoliko različitih tehnoloških platformi koje su postale moguće razvojem molekularne biotehnologije kroz koje desetljeće unatrag. Fast forward u 2021. godinu i možemo reći kako je – iako možda tako ne izgleda – naša pozicija u borbi s virusom ipak osnažena jer konačno imamo cjepivo i to ne jedno već nekoliko njih.
No, ni virus ne miruje, prolaskom kroz veliki broj jedinki optimizira svoje performanse kako bi bio što efikasniji. Evolucijska je strategija virusa (iako je naravno jasno kako se ne radi ni o kakvom promišljenom djelovanju) da se razmnožava u velikim brojevima i da ne kopira suviše precizno svoj genom kako bi se kroz mutacije koje će se u njemu dogoditi možda finim optimiziranjem stvorila njegova što efikasnija verzija odnosno soj. Dio mutacija koje se dogode tijekom njegovih pasaža kroz jedinke ima zapravo neutralni učinak, odnosno ne izazivaju nikakve dramatične promjene u njegovoj građi i funkciji, dio mutacija sigurno i smanjuje njegovu učinkovitost i kao takve se brzo uklanjaju iz bazena kojeg čini populacija nekog virusa. Konačno, dio mutacija čini virus još efikasnijim u procesima koji su bitni za njegov životni ciklus, primjerice da se bolje veže na receptor na stanicama koje inficira. Kako je takav soj efikasniji u pronalasku mjesta za svoje umnažanje jasno je kako s vremenom u situaciji različitih mogućnosti njegovog prijenosa (od nikakvih mjera pa sve do drastičnih lockdowna) postoji veća ili manja šansa da prohara kroz ljudsku populaciju i postane dominantna verzija u odnosu na dosadašnje.
S vremena na vrijeme se u medijima zavrti priča o nekoj od novih varijanti SARS-CoV-2, primjerice od prosinca prošle godine puno se govori o varijanti B.1.1.7. koja je prvotno otkrivena u britanskoj populaciji a danas već postoje podaci o njenoj prisutnosti u gotovo sedamdesetak država. Analizom genoma ovog virusa iz populacije južne Engleske utvrđeno je kako je najveći broj slučajeva pripadao istom filogenetskom klasteru kojeg karakteriziraju multiple mutacije u proteinu spike (delecija 69-70, delecija 144, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H) koje su od posebnog interesa jer su u najbitnijem dijelu virusa, ali i s mutacijama u drugim dijelovima genoma. Preliminarne analize pokazuju kako se ova varijanta širi brže od prijašnjih. Za ovu i prethodne varijante iz Danske, Nizozemske i drugih država utvrđeno je kako ne izaziva agresivniju kliničku sliku, a proizviđači cjepiva su pokazali kako njihova cjepiva induciraju stvaranje protutijela koja su u stanju neutralizirati i te nove varijante virusa. Setom kompleksnih i relativno dugotrajnih pokusa koji ujedinjeno nazivamo metodama reverzne genetike moguće je ispitati u in vitro i in vivo uvjetima kako svaka pojedina virusna mutacija djeluje na prijenos i kliničku sliku (za detaljniji prikaz ove metodologije možete se informirati u ovom članku).
Ipak, varijanta virusa s multiplim mutacijama u spike proteinu nije simpatična te se postavlja pitanje kako je do nje došlo. Prema nekim pretpostavkama moguće je da se radilo o jednom oboljelom s produženim COVID-19 koji je bio imunokompromitiran te je takva produžena infekcija omogućila da virus unutar njega dovoljno dugo živi, evoluira i sakupi mutacije kojima izbjegava imunosni odgovor i to u povećanom obimu nego li je za očekivati u normalnom tijeku bolesti kod pojedinca s adekvatnim imunosnim odgovorom. Druga je mogućnost da je virus u nekom momentu preskočio natrag na životinje (imali smo slučajeve zaraženih životinja na farmama), u njima neko vrijeme evoluirao te se vratio u ljudskog domaćina. Takav je bio slučaj varijante koja je utvrđena na farmi nerčeva u Danskoj a koja je također imala multiple mutacije spike proteina (uključujući i RBD mutataciju Y453F i deleciju 69-70). Drugi set mutacija tog tipa prijavljen je iz Nizozemske. No u Engleskoj nije bilo dokumentiranih slučajeva na uzgojnim farmama pa se ovo objašnjenje čini manje vjerojatnim.
Najsmrtonosniji virusi (poput ebole) većinom nemaju veliki globalni doseg budući da naprosto eliminiraju svoje domaćine prije negoli se uspiju proširiti.
Molekularna evolucija virusa otvara nova pitanja sa svakom novootkrivenom varijantom poput toga hoće li novi soj izazivati teži oblik bolesti ili hoće li cjepivo biti (dovoljno) efikasno. Logika molekularne evolucije s pozicije virusa je da teži bržoj i jačoj propagaciji. Stoga najsmrtonosniji virusi (poput ebole) većinom nemaju veliki globalni doseg budući da naprosto eliminiraju svoje domaćine prije negoli se uspiju proširiti. No niti jedan scenarij ma koliko vjerojatan bio ne možemo unaprijed apsolutno isključiti. S druge pak strane protutijela koja su generirana na protein spike ovog virusa a koja nastaju nakon cijepljenja (ili prebolijevanja bolesti) su zapravo poliklonalna odnosno prepoznaju njegove različite dijelove.
To u osnovi znači da ako se i izmijeni neki dio virusa, još se on ne bi trebao promijeniti do neprepoznatljivosti jer se tada više ne bi mogao vezati na svoj receptor. Poliklonalnost protutijela pak omogućava da ako se to kojim slučajem i dogodi za neki od dijelova proteinske strukture spike proteina, i dalje postoje njeni dijelovi koje naš imunološki sustav može prepoznati. Ipak valja biti na oprezu i stoga je preporuka Svjetske zdravstvene organizacije, a što se također razmatra i na razini EU, da se što je moguće točnije i u realnom vremenu prati molekularna evolucija ovog virusa.
Poznavanje sekvence virusa bitno nam je zbog filogenetskih analiza. Te nam analize koriste kako bismo pomoću sekvenci genoma istražili evolucijske odnose između različitih organizama. Filogenetska stabla su slikovit prikaz „grananja“ odnosno evolucijskog razdvajanja pojedinih organizama. Organizmi na vanjskim granama u takvim prikazima imaju zajedničkog pretka koji se nalazi na grani/čvoru iz kojeg su se dalje odvajale njihove grane. Na temelju filogenetskih analiza izolata virusa kroz vrijeme i u različitim dijelovima svijeta bilo je moguće pratiti njegovu dinamiku širenja i puteve prijenosa. Pojednostavljeno, kada se neka dva soja virusa razdvoje svaki nastavlja neki svoj evolucijski put u kojem skuplja svoj set mutacija te se na temelju razlika u sekvencama sojeva može rekonstruirati kada i gdje u prošlosti su se oni razdvojili (slika 1).
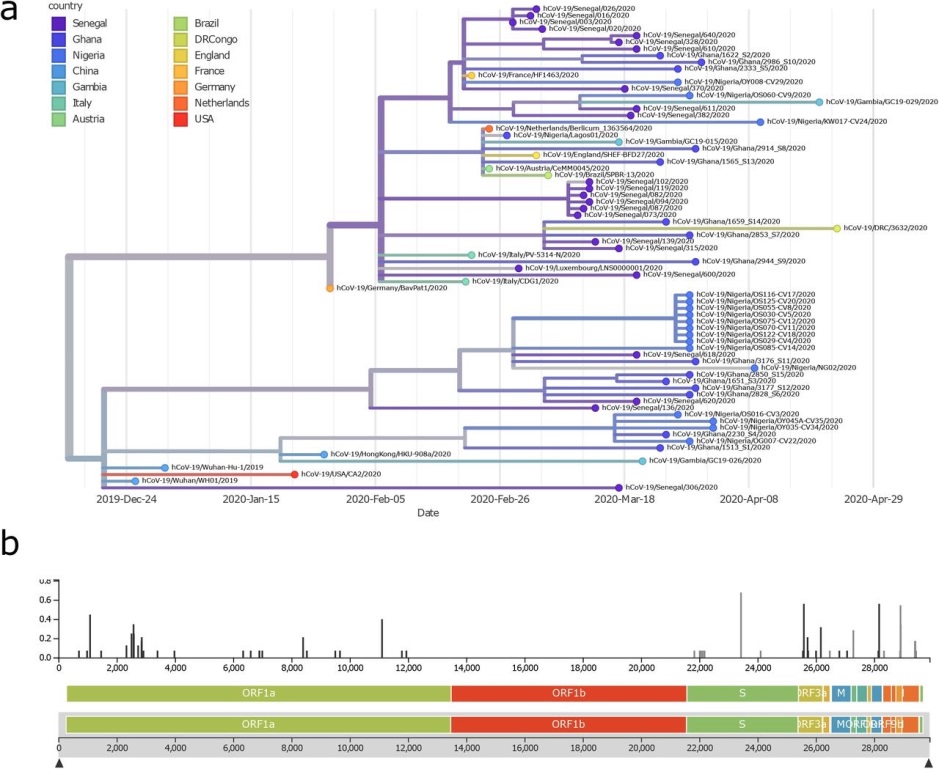
Slika 1. Filogenetsko stablo virusa SARS-CoV-2 u kojem je vidljiva sličnost između varijanti izoliranih u Zapadnoj Africi; Kini te zemljama EU. Preuzeto iz https://www.biorxiv.org/content/10.1101/2020.10.02.323519v1.full
Brzim sekvenciranjem genoma virusa na velikoj skali možemo pratiti njegovu dinamiku i učinkovitost epidemioloških mjera. Ovakav tip sekvenciranja nije do danas bio rutinski primjenjivan u kliničkim laboratorijima te je većinom bio u sferi znanstvenih istraživanja. To je bilo donekle i opravdano jer se svijet preko stotinu godina nije susreo s ovako ozbiljnom pandemijom. Danas je ulaganje u ovakav tip metodologije u okviru translacijske medicine svakako opravdano, no kako se radi o novim tehnologijama, to zahtijeva velike napore u ovladavanju tim metodama kao i nabavu opreme koja je prilično skupa. Rezultati testiranja zatim se analiziraju bionformatičkim algoritmima, što zahtijeva specifičnu edukaciju u području molekularne filogenije koja je prilično dugotrajna te se postavlja pitanje dostupnosti takvog profila stručnjaka.
Primjer zemlje koja je spremno dočekala ovakve izazove je Island. Već sam pisala o njihovoj tvrtki deCODE genetics koja je bila jedan od pionira sekvenciranja humanog genoma te je napravila genomsku banku svojeg stanovništva. Tijekom ove pandemije, gotovo u realnom vremenu, sudjelovala je u sekvenciranju genoma virusa kod svih evidentiranih pacijenata te je na temelju tih podataka utvrđeno 463 varijante virusa koje su prisutne u islandskoj populaciji. Iz ovog je primjera jasno vidljivo koliko je moguće imati virusnih varijanti u jednoj relativno homogenoj i izoliranoj populaciji koja je ispitala detaljno svoj status. Stoga možemo samo pretpostavljati koliko varijanti može postojati unutar država EU a da o svijetu i ne govorimo. Ipak, niste o svima njima bili medijski informirani a vjerojatno i nećete, jer kao što je rečeno, većina tih mutacija ne izaziva ni bržu transmisiju ni težu kliničku sliku.
Već je prije napravljena genomska banka islandskog stanovništva. Sekvenciranjem genoma virusa kod svih evidentiranih pacijenata utvrđene su 463 varijante virusa u islandskoj populaciji. Koliko varijanti može biti u cijeloj EU?
Sekvenciranjem genoma virusa može se pratiti dinamika epidemije, uključujući prostorno-vremensko širenje kao i njegove puteve. Na temelju točnih sekvenci moguće je dizajnirati i po potrebi modificirati dijagnostičke testove, cjepiva i potencijalne lijekove te pratiti kako pojedine mutacije virusa utječu na njihovu efikasnost.
Široku primjenu sekvenciranja omogućio je brzi pad u cijeni kao i povećana brzina obrade uzoraka s jedne strane kao i povećana računalna moć procesiranja dobivenih podataka što nas je uvelo u eru dobivanja informacija o virusu u gotovo realnom vremenu za razliku od prijašnjih mahom retrospektivnih analiza. Za usporedbu s prethodnom epidemijom SARS-a (2002.-2003.) javno su bile objavljene sekvence svega 3 virusna genoma tijekom prvog mjeseca epidemije te se kasnije taj broj povećao na 31 tijekom iduća tri mjeseca. U ovoj epidemiji metagenomskim sekvenciranjem dobiven je prvi genom novog virusa već za nekoliko tjedana, šest genoma objavljeno u siječnju 2020. godine da bi se tijekom prvih 6 mjeseci prošle godine broj objavljenih viralnih genoma popeo na 60 000.
Brzina kojom neki virus mutira određena je biokemijskim svojstvima enzima a radi se zapravo o broju grešaka koje ti (nesavršeno precizni) enzimi generiraju tijekom kopiranja molekule RNA (u slučaju SARS-COV-2 ta molekula je genom virusa) s roditeljskog virusa na njegove potomke. Brzina mutiranja je obično iskazana kao broj mutacija po genomu po replikaciji.
Do brzine kojom neki virus mutira može se doći eksperimentalno. Primjerice možemo sekvencirati neku populaciju virusa te izmjeriti njenu genetičku raznolikost prije i nakon nekog poznatog broja replikacija u laboratorijskim uvjetima. Kako ovaj virus ipak ima neke korektivne mehanizme u replikaciji za razliku od drugih RNA virusa, brzina kojom se stvaraju mutacije srećom nije najbrža moguća koja je viđena za RNA viruse. Kako je rečeno, većina mutacija je štetna za virus te se on nije u stanju dalje replicirati. Samo manji broj mutacija koje daju neku selektivnu evolucijsku prednost virusu bivaju rasprostranjene u populaciji i doprinose brzini kojom on evoluira a koja se iskazuje kao broj supstitutcija po mjestu po godini. Različite evolucijske linije virusa mogu imati različite brzine kojima evoluiraju. Do procjene te brzine se dolazi na temelju sekvenciranja virusnog genoma različitih oboljelih u raznim vremenskim točkama od pojave virusa.
RNA virusi prilično brzo evoluiraju te u prosjeku stječu po jednu genetsku promjenu svakih par dana do par tjedana. Neki od njih stječu genetičke supstitucije gotovo u istom vremenskom tijeku u kojem se odvija prijenos između domaćina. U slučaju SARS-CoV-2 brzina kojom se događaju prijenosi između ljudi je veća od brzine kojom virus stječe supstitucije. Stoga ovaj virus genetičku raznolikost u vidu pojedinih sojeva stječe tijekom tjedana i mjeseci a ne u periodu od nekoliko dana. Taj tijek se može također pratiti sekvenciranjem genoma virusa i time se bavi područje filodinamike. Temeljem ovih analiza moguće je pretpostaviti zajedničkog pretka virusnih genoma koje smo analizirali a koji se tipično nalazi izvan vremenskog prozora u kojem su se počeli uzimati uzorci te se vrijeme kada je on bio prisutan također određuje procjenom i modeliranjem tzv. molekularnog sata odnosno brzine kojom su se odvijale genetske supstitucije u pojedinim granama filogenetskog stabla.
Brzina kojom se događaju prijenosi između ljudi je veća od brzine kojom virus stječe supstitucije. Stoga ovaj virus genetičku raznolikost u vidu pojedinih sojeva stječe tijekom tjedana i mjeseci a ne u periodu od nekoliko dana
Od velike važnosti je utvrditi i kada se SARS-Cov-2 prvi put pojavio u ljudskoj populaciji jer taj podatak ukazuje na to da li se radilo o dugom periodu prijenosa koji je prošao ispod radara prije nego je utvrđen prvi klinički slučaj. Sekvenciranjem genoma virusa iz Wuhana utvrđeno je da su sve pronađene varijante jako bliske te se razlikuju u svega nekoliko nukleotida. Metodom datiranja procijenjeno je kako su sve imale najrecentnijeg zajedničkog pretka koji se pojavio najvjerojatnije u periodu između studenog i prosinca 2019. Iako sekvence genoma virusa iz Wuhana pokazuju relativno malu genetičku raznolikost jasno su vidljive dvije različite filogenetske „loze“ koje ukazuju na separacijski događaj rano u pojavi ovog virusa. To što postoje dvije filogenetske linije ne znači nužno da se one razlikuju u brzini prijenosa ili kliničkoj slici koji izazivaju jer do takvih promjena dolazi obično kasnije u evoluciji virusa kroz stohastičke procese njegovog prijenosa kroz velike popuacije domaćina.
Lock-down nije dakle samo rješavanje akutne situacije već i sprečavanje dugoročnog problema u smislu pojave patogenijeg oblika virusa ili onog na kojeg cjepivo neće više toliko efikasno djelovati. Radi se o utrci između molekularne evolucije virusa i strojeva koji ‘štancaju’ bočice s cjepivom
Stoga i mjere lock-downa ne treba promatrati samo u svjetlu prekidanja lanca prijenosa zarazne bolesti i smirivanja opterećenja na zdravstveni sustav već i kao na metodu kojom se ovi stohastički procesi ipak bar djelomično mogu usporiti u odnosu na situaciju neobuzdanog širenja virusa kroz populaciju. Ovaj segment „usporavanja“ promjena virusa je bitan u uvjetima dok se ne postigne što brža i optimalna procijepljenost populacije do mjere da postaje ograničavajući faktor širenja virusa. Današnji lock-downovi nisu dakle samo rješavanje akutne situacije već i sprečavanje dugoročnog problema u smislu pojave patogenijeg oblika virusa ili onog na kojeg cjepivo neće više toliko efikasno djelovati. Da to slikovito objasnim radi se o utrci između molekularne evolucije virusa (pri čemu imamo sreće jer SARS-CoV-2 nije ni približno toliko brz kao što virusi mogu biti) i velikih bioreaktora i strojeva koji „štancaju“ bočice s cjepivom.
Primjer takvog neobuzdanog širenja i molekularne evolucije virusa je novootkrivena brazilska varijanta koja sadrži ukupno 17 promjena u aminokiselinama te tri delecije. Ova varijanta otkrivena je u 42% uzoraka iz Manausa tijekom prosinca, grada koji se smatra prijestolnicom prirodne okuženosti virusom jer je u njemu oko 75% stanovnika bilo u doticaju s virusom. No varijanta P.1 koja je izolirana u Manausu otvara i dodatna pitanja jer je virus u toj sredini pogodio sličnu konstelaciju mutacija kao i neki virusni sojevi u svijetu s kojima ta populacija nije bila u doticaju. Kada se neka mutacija nađe neovisno (bez direktnog slijeda u filogenetskoj lozi) u raznim krajevima svijeta postaje očito da ona daje neku značajnu evolucijsku prednost virusu i takav nalaz nije bezazlen.

Brazilski grad Manaus smatra se prijestolnicom prirodne prokuženosti. No nova varijanta virusa se neobuzdano širi
Jedno od bitnih pitanja u evoluciji virusa jest identificirati zoonotski rezervoar odnosno životinje od kojih je SARS-CoV-2 virus potekao. Stoga je ovih dana u Kinu došao i tim međunarodnih stručnjaka koji će skupiti uzorke virusa izoliranih iz potencijalnih životinja prijenosnika na lokaciji prve zaraze te ih usmjeriti na filogenetska testiranja.
Poznavanje i praćenje točne sekvence virusa, odnosno identifikacija novih sojeva bitna je i za njegovu dijagnostiku. Metagenomske metode sekvenciranja cijelog genoma nisu rutinski laboratorijski postupci te se za dijagnostičke svrhe koriste validirani testovi koji se rade jednostavnijim metodama. No, treba pratiti evoluciju virusa kako bismo bili sigurni kako naši testovi i dalje jednako efikasno detektiraju i novonastale varijante virusa. Stoga je ovih dana u Njemačkoj a i na razini čitave EU pokrenuta inicijativa za podizanjem i uspostavom kapaciteta za sekvenciranje virusa na razini svih država kako bismo imali prostorno i vremenski što točnije informacije o virusu i da li su naše metode detekcije i dalje dovoljno efikasne. Naravno, ako proces cijepljenja bude trajao duže od najbržeg mogućeg zbog otpora prema cijepljenju, postoji mogućnost da virus mutira u varijante na koje će cjepivo biti manje efikasno, međutim ta efikasnost se neće gubiti drastično i rapidno nego postupno i kroz duže vremensko razdoblje.
Važno je spomenuti kako je sekvenciranje virusa i jedini konačni dokaz da se u nekoga detektirala reinfekcija. Zasada je postotak takovih slučajeva u ukupnom broju oboljelih iznimno mali, no priličan broj tih studija ima reinfekciju potvrđenu samo rutinskom dijagnostikom bez sekvence samog virusa iz prve i ponovljene infekcije. Jedino sekvenca je pravi dokaz de novo reinfekcije jer će se u tom slučaju sekvence virusa iz prve i druge infekcije razlikovati zbog protoka vremena i pasaže kroz populaciju. U slučaju reinfekcije neće se dakle raditi o identičnoj sekvenci te ako se kod oboljelog pronađe ista sekvenca to ukazuje kako je vjerojatno riječ o zalječenju ali ne i izlječenju od prvotne infekcije virusom.

Ako proces cijepljenja bude trajao duže zbog otpora prema cijepljenju, postoji mogućnost da virus mutira u varijante na koje će cjepivo biti manje efikasno, ali se efikasnost se neće gubiti drastično i rapidno nego postupno i kroz duže razdoblje. Prvo cijepljenje protiv covid-19 u Hrvatskoj (screenshot N1)
Konačno, ako i kada jednoga dana možda bude registriran i kakav ciljani lijek za SARS-Cov-2 također će biti bitno pratiti da li takav lijek dijeluje na najnovije varijante virusa.
Ovakav napredak u molekularnoj virologiji, i razvoju novih platformi cjepiva koji je nažalost isforsiran globalnom epidemijom SARS-CoV-2, zapravo je dobra stvar jer nas u našoj globalizacijskoj stvarnosti takve epidemije, s možda još pogubnijim posljedicama, tek čekaju. Protekle godine učinjeni su znanstveni iskoraci za koje bi u nekim normalnim vremenima trebalo možda i cijelo desetljeće, ne zato jer se sada jurilo i površno radilo nego stoga jer je u ovu sferu istraživanja uloženo novaca koliko nije cijelo prethodno desetljeće, primjerice, samo u razvoj cjepiva uloženo je nekoliko milijardi dolara. Svi znamo kako ide ona poslovica o novcu i muzici. I iako je ne možemo primijeniti kao apsolutni zakon, sasvim je sigurno da u uvjetima enormnog interesa za ta područja istraživanja koje prati i enormno financiranje, iskoraci postaju realitet. Znanost je u najkraćem mogućem roku koji je danas nama tehnološki dostupan dala rješenja za niz pitanja u ovoj epidemiji i sad je na nama da prigrlimo blagodati ovakvog razvoja i cijepimo se u što većem broju što je najprije moguće kako bismo prerezali lanac širenja ove bolesti.
1)WHO: Genomic sequencing of SARS-CoV-2 A guide to implementation for maximum impact on public health. 8 January 2021. https://www.who.int/publications/i/item/9789240018440
2) European centre for disease prevention and control. Sequencing of SARS-CoV-2. 18 January 2021. https://www.ecdc.europa.eu/en/publications-data/sequencing-sars-cov-2
3) European centre for disease prevention and control. Rapid increase of a SARS-CoV-2 variant with multiple spike protein mutations observed in the United Kingdom. 20 December 2020. https://www.ecdc.europa.eu/en/publications-data/threat-assessment-brief-rapid-increase-sars-cov-2-variant-united-kingdom



