I studenti s malo novca mogu biti vrhunski znanstvenici: Quo vadis, chemia?
Razvoj računala je omogućio kemiji da prestane biti znanost kojoj je mjesto isključivo u laboratoriju – danas, računalna kemija služi ne samo kao teorijska podloga eksperimentalnoj kemiji, već i preuzima dio njenih poslova. A uz nezaustavljiv rast računalne snage, igralište za in silico istraživanja ubrzano se širi.
Razvoj računala je omogućio kemiji da prestane biti znanost kojoj je mjesto isključivo u laboratoriju – danas, računalna kemija služi ne samo kao teorijska podloga eksperimentalnoj kemiji, već i preuzima dio njenih poslova. A uz nezaustavljiv rast računalne snage, igralište za in silico istraživanja ubrzano se širi.
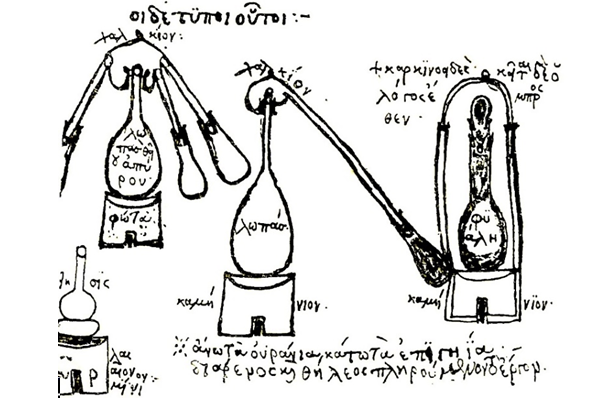
Od svojih prapočetaka u alkemiji i metalurgiji, kemija je bila neraskidivo vezana uz rad u laboratoriju. Prije više od dvije tisuće godina, grčki i egipatski alkemičari su destilirali ulja i kiseline, pirolizirali drvo i žarili rudaču u visokim pećima. Kao i današnji kemičari, svoja opažanja su zapisivali, a naučena znanja prenosili budućim generacijama.
Zosimova aparatura za destilaciju (oko 300. pr. Kr.). Današnje aparature nisu puno drugačije.
Kemija se rješava misticizma i pretvara u modernu znanost tek krajem 18. stoljeća, poglavito zaslugama Antoinea Lavoisiera (teorija gorenja i zakon očuvanja mase) i Johna Daltona (atomistička teorija). Kroz 19. stoljeće njen razvoj se ubrzano nastavlja – sintetiziraju se novi spojevi i razvijaju eksperimentalne tehnike. Nastaju i korisne kemijske teorije: 1832. Wöhler i Liebig iznose teoriju da fukcionalne skupine određuju reaktivnost organskih spojeva, a 1869. Mendeljejev predstavlja periodni sustav elemenata. Te teorije, iako uspješne u predviđaju ishoda eksperimenata, i dalje kemiji ne nude čvrstu konceptualnu podlogu niti duboke temelje.
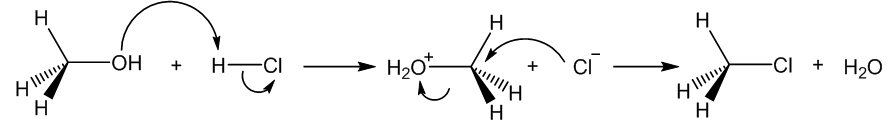
Takva podloga nastaje tek razvojem kvantne teorije u prvoj polovici 20. stoljeća. Robinson i Ingold, potaknuti saznanjima o elektronskoj strukturi atoma, tada razvijaju teoriju da se kemijeske reakcije sastoje od niza koraka, u kojima se događaju točno određeni pomaci atoma. Nešto kasnije, Woodward i Hoffmann uspješno primjenjuju koncepte proizašle iz kvantne mehanike (molekularne orbitale) za objašnjenju jedne neobične klase reakcija (pericikličkih reakcija).
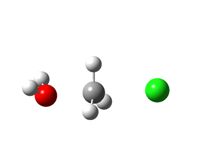
Reakcijski mehanizam zamjene (supstitucije) hidroksilne skupine kloridom. Zaobljene strelice predstavljaju parove elektrona koji omogućuju napad, odnosno izlazak pojedinih skupina iz molekule. Robinson je predložio ovakav mehanizam na temelju rezultata eksperimenata vezanih uz brzine reakcija, više od pola stoljeća prije nego što je on potvrđen računalnom kemijom.
Cilj tih teorija bio je povezati rezultate kvantne mehanike koji su se odnosili na individualne atome i male molekule, s opažanjima u laboratoriju koja opisuju prosječno ponašanje ogromnog mnoštva (često velikih) molekula. No, veza je bila tek kvalitativna – za izravniju primjenu kvantne mehanike na složene kemijske sustave i kvantitativno slaganje eksperimenta i teorije trebalo još malo pričekati.
Ideja računalne kemije
Još 1888. godine Gay-Lussac je izjavio kako vrijeme kada ćemo većinu kemijskih fenomena moći izračunati vjerojatno nije daleko.[1] Oko pola stoljeća kasnije, R. Feynman u svom je stilu komentirao kako se sve što žive stvari rade može razumijeti preko mrdanja i vrpoljenja atoma.[2] No, Feynmanov jiggling and wiggling atoma matematički je vrlo kompliciran – Paul Dirac 1929. piše:
Fizikalni zakoni potrebni za matematičku teoriju velikog dijela fizike i cijele kemije potpuno su poznati, a jedini je problem što točna primjena tih zakona dovodi do jednadžba koje su prekomplicirane da bi bile rješive. Zbog toga, poželjno je razviti praktične približne metode primjene kvantne mehanike, koje mogu dovesti do objašnjenja osnovnih svojstava kompleksnih atomskih sustava bez puno računanja.[3]
U razvoju „praktičnih približnih metoda“ koje je spominjao Dirac sudjelovali su mnogi znanstvenici tijekom druge polovice 20. stoljeća, a najuspješniji među njima bili su Pople i Kohn, koji su 1998. dobili Nobelovu nagradu za razvoj računalne kemije. Izazova je bilo mnogo – kvantne mehanike trebalo je pojednostaviti tako da postanu rješive, a zatim je svu tu matematiku trebalo pretvoriti u jezik razumljiv elektroničkim računalima, koja su se počela pojavljivati u to doba (ENIAC je sastavljen 1946.). Istovremeno, uvedena pojednostavljenja nisu smjele biti pregruba jer bi to rezultiralo u slaboj točnosti izračuna.
Međutim, ideja primjene kvantne mehanike u kompliciranim kemijskim sustavima bila je jako privlačna ne samo zato jer je kemiji davala čvrsto i stabilno konceptualno uporište, već i zato što je kemiju približila subjektu svog istraživanja. Nasuprot zaključivanja na temelju makroskopskih laboratorijskih eksperimenata, računalna kemija nudi ulazak u svijet molekula i neposredno promatranje njihovog plesa, pretvorbi i interakcija.
Izračunata prijelazna struktura za reakciju prikazanu na prethodnoj slici, koja potvrđuje Ingoldov mehanizam (vodik je označen bijelo, kisik crveno, klor zeleno, a ugljik sivo).
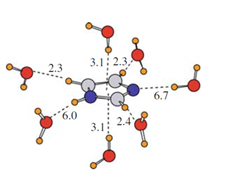
Energije vodikovih veza u imidazolu. Rezultati izračuna sugeriraju kako su vodikove veze s dušikom (označenim plavo) otprilike dvostruko jače od svih ostalih, što nije lako odrediti bez računa.[4]
Danas, računalna se kemija od poveznice s fizikom pretvorila u praktički ravnopravan komplement klasičnoj laboratorijskoj kemiji. Ona i dalje ima jedinstvenu ulogu u objašnjavaju interakcija među atomima, no koristi se i za planiranje sinteze (hoće li neka reakcija „ići“ ili ne?), interpretaciju eksperimentalnih rezultata (je li neka promjena predviđena računom uočena i u eksperimentu?) i predviđanje svojstava novih, još nesintetiziranih spojeva. Dobar dio uspjeha duguje brzom razvoju računala, koji je omogućio povećanje i točnosti i veličine proučavanog sustava.
Računalna kemija daje mogućnost promatranja individualnih mikroskopskih procesa vezanih uz otapanje soli, kao što su narušavanje kristalne rešetke i hidratacija pojedinačnih iona.
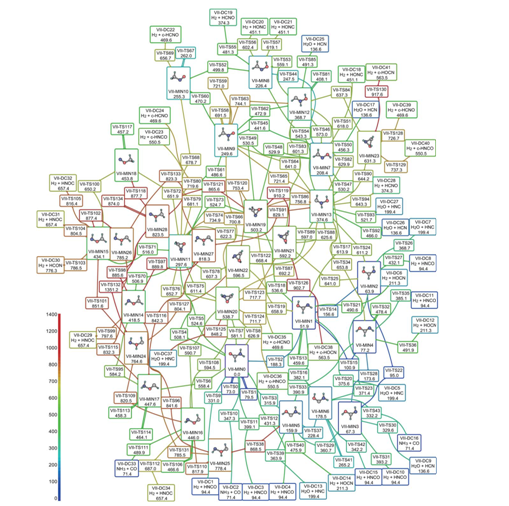
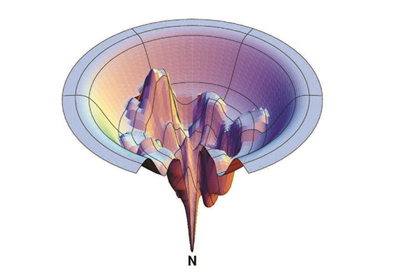
Ploha potencijalne energije za CH3NO, koja prikazuje sve moguće stabilne molekule koje mogu nastati kombinacijom tih atoma. Boja odgovara energiji – za prijeći plavim putem potrebno je manje energije nego za crveni. Prema tome, slika prikazuje koje su od stotina zamislivih reakcija nemoguće, koje su vjerojatno izvedive, a koje potencijalno opasne (jer u njima na primjer nastaje vodik uz oslobađanje energije). [5]
Razvoj računala i Mooreov zakon
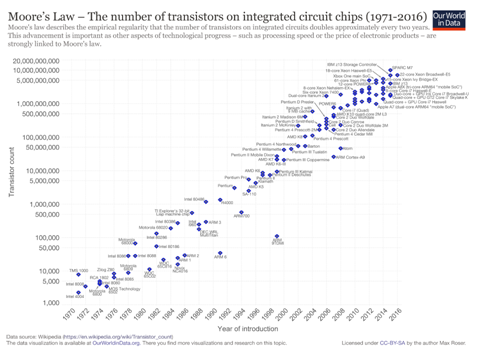
U poznatom intervjuu iz 1965., Gordon Moore predvidio je kako će broj komponenata po čipu sljedećih nekoliko godina povećavati ugrubo eksponencijalno. To predviđanje pokazalo se nevjerojatno točnim – računalna snaga i danas raste eksponecijalno, čak i kada se cijena uzme u obzir.
Grafički prikaz Mooreovog zakona. Na Y osi je broj tranzistora u eksponencijalnoj skali, dok X os prikazuje vrijeme.
U dubinu i širinu
Tako duga valjanost Mooreovog zakona imala je vrlo blagotvoran učinak na sve znanstvene discipline koje se oslanjaju na računala, pa tako i na računalnu kemiju. Snažnija računala potaknula su razvoj naprednijih metoda, koje koriste jako malo aproksimacija te daju vrlo točne rezultate. Jedan gostujući profesor, koji je dugi niz godina radio za Pfizer, tijekom predavanja o dizajnu lijekova ispričao je anegdotu kako im američka FDA zbog jednog minijaturnog signala u NMR spektru nije htjela odobriti lijek, tvrdeći kako se ne radi o čistoj supstanciji već o smjesi. Računalnom kemijom njegova je grupa uspjela dokazati postojanje tog signala u čistom lijeku, što im je u konačnici donijelo odobrenje. Cijela priča postaje zanimljivija kada se stavi u financijski kontekst – prodaja tog lijeka bila je procijenjena na oko 5 milijuna dolara dnevno.
Osim za vrlo točne kvantno-mehaničke račune vezane uz relativno male molekule, snažna računala mogu se iskoristiti i za modele s daleko grubljim aproksimacijama, koji ne koriste kvantnu, već klasičnu mehaniku pa atome promatraju kao točkaste mase povezane oprugama. Takvi su modeli manje računalno zahtjevni, pa se mogu koristiti za modeliranje velikih bioloških sustava kao što su proteini, dijelovi molekule DNA, citoplazme ili stanične membrane. Simulacije takvih proporcija otvaraju prozorčić prema promatranju života stanice te utiru put ka području računalne biologije.
Interakcija vezikule (omotača koji stanice koriste za pohranu i transport tvari) i stanične membrane u izdanju Sciencea posvećenom računalnoj kemiji.[6] Obratite pozornost na godinu (2008.) i usporedite s prethodnom slikom – danas imamo barem 50 puta jača računala od onih potrebnih za stvaranje ove slike.
Prikaz vrlo napučenog i živahnog proteinskog svijeta koji se nalazi unutar svake stanice.
I dok se izuzetno snažna i skupa računala mogu iskoristiti za odličnu znanost, čak i cijena „dovoljno dobrih“ računala je takva da su ona postala dostupna gotovo svima. Tako praktički svi današnji studenti kemije imaju pristup instrumentima računalne kemije (bilo u vidu laptopa ili kompjutora u računalnoj učionici), što se za ostale moderne kemijske instrumente ne može reći – snažni laseri, veliki magneti hlađeni tekućim dušikom i izvori rendgenskih zraka daleko su skuplji, i time manje dostupni slabije financiranim institucijama (a ako ih i imaju, studenti će teško stići na red).
Smatanje proteina u pogrešnu konfiguraciju dovodi do narušavanja njegove biološke funkcije, što može biti uzrokom različitih bolesti. Uz pomoć milijuna računala diljem svijeta, Folding at home objavio je niz radova vezanih uz razne teme, od Alzheimerove i Parkinsonove bolesti, tumora i proteina p53 do viralnih infekcija
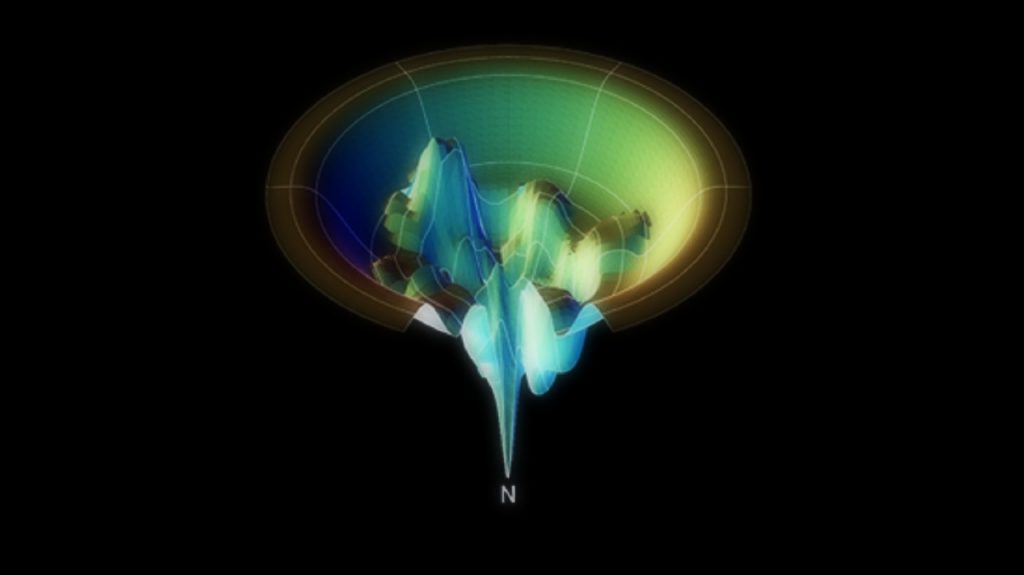
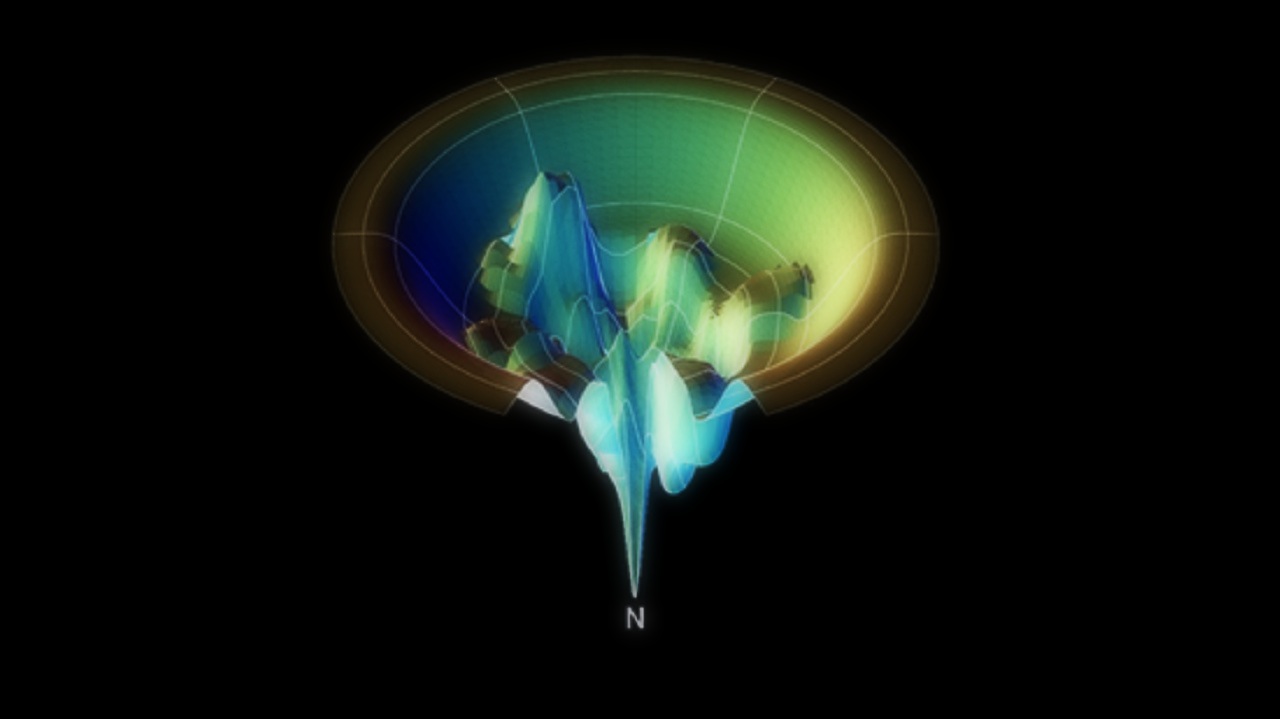
Činjenicu da danas svatko posjeduje računalo odlično je iskoristio projekt Folding at home sa Stanforda, koji je omogućio svima zainteresiranima da koriste svoje kućno računalo, a kasnije i Playstation, pa čak i mobitel (!) za modeliranje smatanja proteina. Proteini (bjelančevine) su biološki izuzetno važne makromolekule koje se sastoje od nekoliko stotina ili tisuća aminokiselina povezanih u lanac koji je smotan na točno određen način. Smatanje proteina u pogrešnu konfiguraciju dovodi do narušavanja njegove biološke funkcije, što može biti uzrokom različitih bolesti. Uz pomoć milijuna računala diljem svijeta, Folding at home projekt objavio je niz radova vezanih uz razne teme, od Alzheimerove i Parkinsonove bolesti, tumora i proteina p53 do viralnih infekcija, malarije i dijabetesa, a sve pod zajedničkim nazivnikom smatanja proteina.[7]
Energetski profil smatanja proteina. U pravilu, protein će završiti u energetski najpovoljnijoj konfiguraciji, označenoj N, no mutacije ili druge molekule mogu uzrokovati pogrešno smatanje.
Simulacija nukleosoma – „paketa“ u kojem se nalazi DNA. Drugi dio videa prikazuje interakciju DNA i proteina p53, čija je glavna uloga zaštita organizma od nastanka tumora.
Računalom potpomognuti dizajn
Računala su se pokazala osobito korisnima u big data područjima, gdje postoji ogroman broj „kandidata“ među kojima treba odabrati najbolje. U dizajnu lijekova i kemiji materijala, za takav posao često se koriste QSAR (quantitative structure-activity relationship) metode. One iz strukture ili sastava (structure) neke molekule ili materijala statističkim metodama pokušavaju procijeniti njihova svojstva (activity), uspoređivanjem kandidata s bazom podataka već poznatih molekula, odnosno materijala.
Pomoću QSAR metoda tako je moguće relativno brzo pročešljati bazu podataka s desecima tisuća kandidata i pronaći recimo stotinu obećavajućih, koje se onda može ili dalje istraživati drugim računalnim metodama ili sintetizirati. U takvim slučajevima, upotreba računala štedi vrijeme i novac, a i ekološki je daleko prihvatljivija – jedina „kemikalija“ koja se troši je struja.
Konkretno, QSAR metode popraćene kvantno-mehaničkim računima koriste se u objašnjenju svojstava i dizajnu novih magneto-električkih materijala, a u nedavnim radovima počinju se upotrebljavati i metode strojnog učenja.[8] U još jednom radu nedavno objavljenom u Natureu, istraživači su računalnim metodama uspjeli povezati makroskopska mehanička svojstva metalnog stakla (materijala velike čvrsoće koji su otporni na pucanje i koroziju) s interakcijama između atoma metala na mikroskopskoj razini.[9] Takvi rezultati otvaraju mogućnost dizajna materijala kojima ćemo moći odabrati svojstva i zatim ih pokušati „pronaći“ računalom, bez paljenja visokih peći.
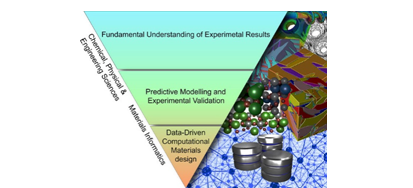
Tri uloge računalnog modeliranja u dizajnu magneto-električnih materijala.8 Gore – fundamentalno razumijevanje eksperimentalnih rezultata; sredina – predviđanje putem modeliranja i validacija eksperimenata; dolje – primjena statističkih (QSAR) tehnika za dizajn materijala.
Lijekovi računalom
U dizajnu lijekova, dva osnovna problema su pronaći molekulu (supstrat) koja se što bolje, ali i selektivnije veže za aktivno mjesto neke biološke makromolekule, u pravilu proteina. Dobro vezanje povećava djelotvornost lijeka i smanjuje aktivnu dozu, što olakšava posao metabolizmu. S druge strane, selektivnost umanjuje mogućnost nuspojava (selektivan supstrat vezat će se samo za jedno aktivno mjesto i učiniti samo jednu stvar u organizmu). QSAR metode koriste se za rješavanje oba problema, ali i za procjenu topljivosti i metabolizma lijeka (hoće li se supstrat i njegovi metaboliti zadržati u mastima, ili se brzo izlučiti urinom?).
Primjer računalom potpomognutog dizajna lijekova. U prvoj polovici videa (0:18–0:35) prikazano je grubo traženje dobrog kandidata za supstrat, koji se zatim u drugoj polovici optimizira sitnim promjenama.
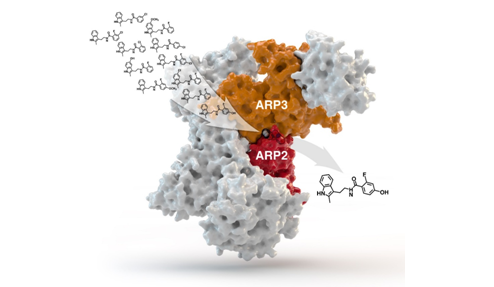
Efikasan inhibitor proteinskog kompleksa Arp2/3 pronađen je višerazinskim metodama iz niza kandidata. [10] Kompleks Arp2/3 igra važnu ulogu u kretanju stanica, pa njegova inhibicija sprječava metastaziranje stanica raka.
Posljednji veliki korak naprijed u računalom potpomognutom dizajnu lijekova bio je razvoj višerazinskih (QM/MM) računalnih modela za proučavanje velikih sustava (Nobelova nagrada 2013.). Ideja iza takvih modela je podijeliti modelirani sustav (na primjer, cijeli protein sa supstratom u aktivnom mjestu) na više dijelova. Važniji dijelovi sustava (aktivno mjesto i supstrat) onda se modeliraju točnijim (kvantno-mehaničkim, QM) metodama, dok se daleko veći, ali manje važan ostatak modelira jednostavnijim (MM) metodama. QM/MM metode danas se koriste diljem svijeta, pa tako i u Hrvatskoj – slika ispod ovog teksta nastala je nakon višemjesečnog računanja na Institutu Ruđer Bošković.
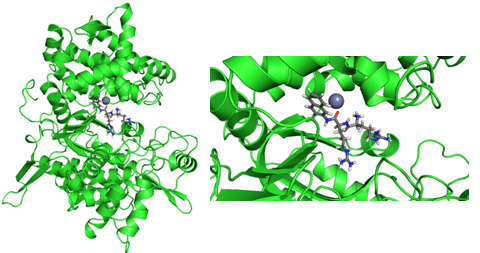
Lijevo: Protein DPP III s Arg2-naftil-2-amid (RRNA) supstratom u aktivnom mjestu. Desno: uvećana slika aktivnog mjesta. RRNA je jako dobar supstrat za DPP III, pa se može koristiti za ispitivanje njegovih mutanata (koje mutacije u genetskom kodu utječu na aktivnost DPP III, a koje nisu važne?).
Kada je prije oko 50 godina čovjek sletio na mjesec, mnogi su bili uvjereni kako nam slijedi doba istraživanja svemira. To se baš i nije ostvarilo, no umjesto plovidbe svemirom dobili smo mogućnost krstarenja nanosvijetom atoma i molekula, i to brodovima koje nosimo u torbama i džepovima. Ideja da se makrokozmos („veliki svijet“) zrcali u mikrokozmosu („malom svijetu“) proganjala je brojne alkemičare, teologe i filozofe da bi se konačno ostvarila u svijetu znanosti.
Igor Rončević radi kao poslijedoktorand na Prirodoslovno-matematičkom fakultetu u Zagrebu.
[1]We are perhaps not far removed from the time when we shall be able to submit the bulk of chemical phenomena to calculation.
[2]…everything that living things do can be understood in terms of the jigglings and wigglings of atoms. Sličan je stav dijelio i Tit Lukrecije Kar (1. st. pr. Kr.), koji piše: The atoms are eternal and always moving. Everything comes into existence simply because of the random movement of atoms, which, given enough time, will form and reform, constantly experimenting with different configurations of matter from which will eventually emerge everything we know…
[3]The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble. It therefore becomes desirable that approximate practical methods of applying quantum mechanics should be developed, which can lead to an explanation of the main features of complex atomic systems without too much computation.
[4] T. K. Schneider, Int. J. Quantum Chem., 2006, 106, 843-851.
[5] S. Maeda, Y. Harabuchi, Y. Ono, T. Taketsugu, K. Morokuma. Int. J. Quantum Chem. 2015, 115, 258–269. DOI: 10.1002/qua.24757
[6] Izvor slike: Wataru Shinoda/Research Institute for Computational Sciences, National Institute of Advanced Industrial Science and Technology, Japan; Mike Klein/Center for Molecular Modeling, University of Pennsylvania
[7] Više informacija na http://folding.stanford.edu/diseases/
Radovi nagrađeni ovogodišnjim Nobelom za fiziku nisu samo riješili zagonetku mosta između mikro i makro svijeta, oni su napravili i praktične alate za gradnju kvantnih tehnologija, piše Davor Horvatić. Njihova najvažnija poruka nije da je „kvantno moguće i u velikim makroskopskim sustavima“, nego da je priroda dosljedna.
Kada brojevi postaju preveliki, brojanje postaje aproksimativno, bez obzira radi li se o životinjama ili ljudima, ili o realnim brojevima zapisanim u arhitekturi računala, piše Aleksandar Hatzivelkos. Nedavno otkriveni CVS algoritam omogućava jednostavnu no dosta preciznu aproskimaciju, uz minimalno korištenje resursa.
Višeagentni modeli, od Schellingovih novčića do MATSim simulacija na računalnim oblacima, pokazuju kako se kompleksni društveni obrasci mogu 'uzgojiti' iz jednostavnih pravila, piše Slaven Smojver. Omogućavaju simuliranje emergentnih ponašanja, a najznačajnija prednost i korist višeagentnih modela je mogućnost izvođenja eksperimenata koje u stvarnosti ne bismo mogli, smjeli ili znali provesti.
Koristimo kolačiće kako bismo osigurali pravilno funkcioniranje web-stranice, značajke društvenih mreža te analizirali promet i performanse.
Funkcionalni kolačići
Uvijek aktivni
Ovi kolačići su nužni za funkcioniranje web stranice i ne mogu se isključiti u našim sustavima. Možete postaviti svoj preglednik da blokira ove kolačiće ili da vas o njima obavijesti, ali neki dijelovi web stranice tada možda neće raditi.
Preferences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistički kolačići
The technical storage or access that is used exclusively for statistical purposes.Ovi kolačići omogućuju nam analizu posjeta i izvora prometa kako bismo mogli mjeriti i poboljšati učinkovitost naše web stranice. Pomažu nam saznati koje su stranice najpopularnije, a koje najmanje, te kako se posjetitelji kreću po web stranici.
Marketinški kolačići
Tehnička pohrana ili pristup potrebni su za izradu korisničkih profila za slanje oglasa ili za praćenje korisnika na web stranici ili na nekoliko web stranica u slične marketinške svrhe.