MOLEKULARNA BIOLOGIJA
Iskoraci u liječenju rijetkih bolesti i Down sindroma. Terapije tehnologijom CRISPR-Cas9
 Tamara Čačev / 30. srpnja 2025. / Perspektive / čita se 11 minuta
Tamara Čačev / 30. srpnja 2025. / Perspektive / čita se 11 minuta

Tamara Čačev / 30. srpnja 2025. / Perspektive / čita se 11 minuta
Do sada je zabilježeno više od 7.000 rijetkih bolesti, od kojih danas boluje preko 300 milijuna ljudi. Tu nije riječ o zanemarivom problemu, piše Tamara Čačev, no većina tih bolesti u svojoj osnovi ima promjenu u nekom od gena, te ih njihove kliničke i genetske razlike čine jednim od najizazovnijih područja u suvremenoj medicini.
Pod pojmom rijetkih bolesti, ili u engleskom govornom području tzv. orphan diseases, podrazumijevamo heterogenu skupinu bolesti niske prevalencije (manje od 1 oboljelog na 2.000 stanovnika) u čijoj osnovi se većinom nalazi promjena u nekom od gena čija se funkcija najčešće tako i otkrije. No, takvih rijetkih, mogli bismo čak reći i individualnih promjena u genomu je puno, pa stoga baze podataka poput Orphaneta bilježe već preko 7.000 različitih upisa rijetkih bolesti od kojih danas boluje preko 300 milijuna ljudi. Iz ovih je podataka jasno kako se zapravo ipak ne radi o zanemarivom javno zdravstvenom problemu, no klinička i genetska heterogenost rijetkih bolesti čini ih jednim od najizazovnijih područja u suvremenoj medicini.
Istraživanje njihove genetske osnove pruža jedinstven uvid u temeljne mehanizme molekularne biologije čovjeka kroz rijetku priliku istraživanja svega što može poći krivo ukoliko neki gen ne izvršava svoje funkcije onako kako bi trebao. Kako ne bi ostalo sve apstraktno, navest ću neke primjere rijetkih bolesti koje su možda najviše doprle do šire javnosti. Jedan od takvih primjera je bolest „djece leptira“ – bulozna epidermoliza, za koju je identificirano više gena te njihova funkcija. Druga vjerojatno također široj javnosti poznata rijetka bolest je progerija (Hutchinson-Gilford sindrom), bolest uzrokovana mutacijom u genu LMNA koja je pružila uvid u mehanizme starenja. Naime, oboljeli stare puno brže nego što je to uobičajeno pa u tinejđerskoj dobi već izgledaju kao da su u dubokoj starosti što se nažalost reflektira i na njihov zdravstveni status i životni vijek.
Bolesti za koje su možda čuli upućeniji čitatelji poput ataksije-telangiektazije (uzrok je mutacija u genu ATM) i Fanconijeva anemija omogućile su uvid u mehanizme popravka DNA ali i karcinogeneze dok je Tangierova bolest (mutacije gena ABCA1) dala uvid u metabolizam kolesterola i patogenezu ateroskleroze. Stoga možemo reći kako su mnogi od temeljnih mehanizama molekularne biologije čovjeka otkriveni su upravo kroz istraživanje ovih bolesti.
Donedavno se dijagnostika ovih bolesti temeljila na anamnezi vrsnih pedijatara koji su u vrlo kratkom vremenu (radi se nekad i o satima) morali na temelju kliničke slike odlučiti o kojem bi se genu eventualno moglo raditi te bi se onda dijete i testiralo na funkcionalnost toga gena. Srećom, razvoj next generation sequencing (NGS) metoda omogućio je da se u kratkom vremenu može sekvencirati čitav genom pa tako danas barem imamo sredstvo koje možemo upotrijebiti da saznamo što se nalazi u podlozi neke neobjašnjive kliničke prezentacije koja bi mogla biti dio nekog genetskog poremećaja. Međutim, interpretacija podataka ostaje izazov. Studije pokazuju da se u 25–50 % slučajeva otkrije uzročna varijanta, dok su ostale varijante klasificirane kao VUS (variant of unknown significance), termin pod kojim podrazumijevamo promjenu u genomu čiji utjecaj na kliničku sliku još nije razjašnjen. Jasno je da je također potrebna i sveobuhvatna multidisciplinarna analiza koja će povezati kliničku genetiku, molekularn biologiju, bioinformatiku, te baze podataka humanog genoma koje tek trebamo napuniti relevantnim podacima kako bi nam služili u budućim slučajevima.
No, što je s lijekovima koji bi trebali također proizaći iz ovakvih istraživanja? Ovdje dolazimo do glavne prepreke koja se sve donedavno činila nepremostivom iz više razloga. Naime, čak i kada znamo, što kako smo prethodno rekli počesto i nije slučaj kada se rodi dijete s nekom rijetkom bolešću, o kojem se točno poremećaju gena se radi, terapija, osim u malom broju nekih metaboličkih bolesti, nije dostupna. Naprosto nisu pronađena rješenja kako nadomjestiti sve funkcije nekog gena u organizmu kroz sve životne faze. Ako dijete ima sreće pa je zahvaćen gen koji nije od esencijalne važnosti za neposredno preživljavanje, tada započinje doživotna borba s bolesti kojoj mali broj usko specijaliziranih liječnika zna kako pristupiti. Često se u podizanju svijesti o ovim bolestima spominje i neisplativost istraživanja i pronalaska lijekova za tako mali broj individualnih korisnika te stoga i nepostojanje interesa od strane farmaceutske industrije da se više angažira na ovom području.
Napredak molekularne biologije omogućio da rijetke bolesti danas postanu područje najbrže rastućih inovacija
Ipak povod za pisanje ovog teksta su nedavni ogromni iskoraci na ovom području jer je napredak molekularne biologije omogućio da rijetke bolesti postanu područje najbrže rastućih inovacija. Danas se razvijaju genske terapije, RNA terapije, enzimske nadomjesne terapije i terapije temeljene na antisense oligonukleotidima. Javnosti je vjerojatno najpoznatija terapija Spinrazom (nusinersen) – terapija temeljena na antisense nukleotidima koja mijenja alternativno izrezivanje eksona gena SMN2 kod spinalne mišićne atrofije o čemu sam već pisala u prethodnim tekstovima. Drugi lijek Zolgensma, a riječ je o vektoru koji unosi funkcionalnu kopiju SMN1 gena u stanicu također je bio u žiži medijskog interesa i u našoj javnosti. Revolucionarne uspjehe koje takva terapija može imati, zasjenila je njena milijunska cijena popraćena razumljivom željom roditelja da njihova djeca tu terapiju dobiju iako možda i nisu pravi kandidati za nju.
Najnoviji uspjeh na ovom području napravili su američki znanstvenici koji su prvi u svijetu primijenili personaliziranu gensku terapiju za liječenje bebe rođene s po život opasnom i neizlječivom genetskom bolešću, rijetkim poremećajem zvanim deficit karbamoil-fosfat-sintetaze 1 (CPS1) koji se javlja s incidencijom od 1 na 1.3 milijuna novorođenčadi. Deficit CPS1 je metabolički poremećaj u kojem jetra nije sposobna razgraditi nusprodukte metabolizma proteina što dovodi do toksičnog nakupljanja amonijaka u tijelu. To može izazvati teška oštećenja mozga i jetre. Standardno liječenje uključuje dijetu s vrlo niskim udjelom proteina, dok se ne steknu uvjeti za transplantaciju jetre. Međutim, tijekom tog razdoblja postoji stalni rizik od naglog zatajenja organa uslijed infekcije, traume ili dehidracije. Visoke razine amonijaka mogu izazvati komu, edem mozga, trajno neurološko oštećenje, pa čak i smrt. Tim znanstvenika s Dječje bolnice u Philadelphiji (CHOP) i Perelmanove medicinske škole Sveučilišta Pennsylvania razvio je ciljanu terapiju upravo za ovo dijete pomoću tehnologije za uređivanje gena CRISPR-Cas9. U istraživanju su sudjelovali i Acuitas Therapeutics, Integrated DNA Technologies, Aldevron i Danaher Corporation.
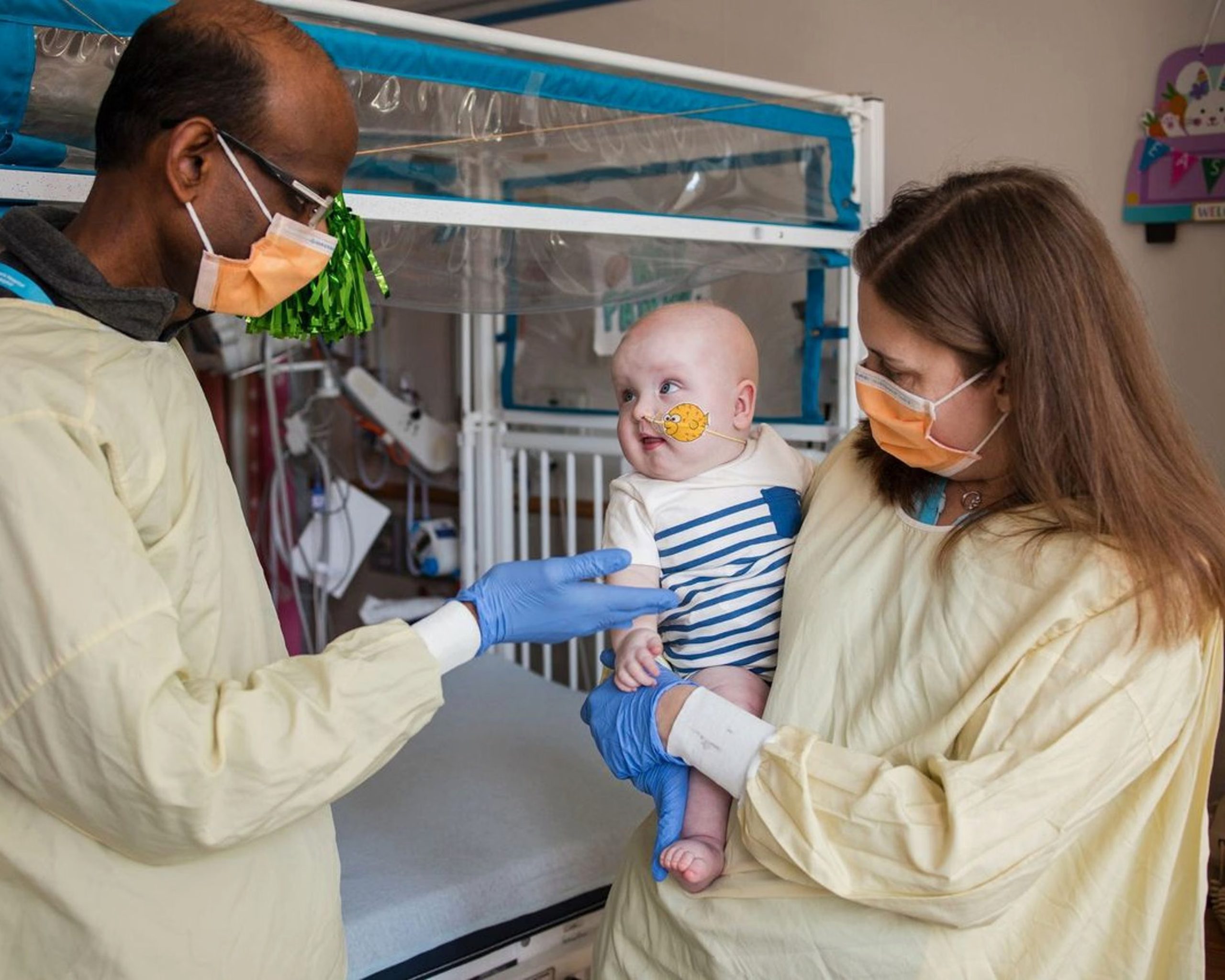
Riječ je o prvom poznatom slučaju primjene personalizirane CRISPR-terapije kreirane namjenski za jednog pacijenta. Koristeći tehnologiju CRISPR, ispravili su mutaciju ovog gena u jetrenim stanicama djeteta koja je bila odgovorna za poremećaj. Dijete je u dobi od šest mjeseci prvo primilo vrlo nisku dozu genske terapije, te potom višu dozu. Istraživači su primijetili znakove pozitivnog odgovora budući da je dijete moglo podnijeti veću količinu proteina u prehrani te je smanjena doza lijekova potrebnih za održavanje normalnih razina amonijaka. Iako je terapija još u fazi praćenja, dijete sad ima 10 mjeseci i mnogo toga ostaje nepoznato, istraživači su oprezno optimistični u vezi s daljnjim napretkom djeteta. Rezultati su predstavljeni na godišnjem sastanku Američkog društva za gensku i staničnu terapiju (ASGCT) u svibnju ove godine, a studija je objavljena u časopisu The New England Journal of Medicine.
Proces od dijagnoze do terapije trajao je svega šest mjeseci, što je nevjerojatno brzo pogotovo kada to usporedimo s konvencionalnim načinom razvoja lijekova. Ipak, proces stvaranja ovakvih terapija je i dalje skup, iako se naglašava kako je malo skuplji od transplantacije jetre koja bi ovom djetetu trebala. Stoga je bitno naglasiti kako je korištena tehnologija koja se temelji na platformi koja se može prilagoditi za liječenje različitih genetskih bolesti, otvarajući mogućnost razvoja i drugih personaliziranih terapija. Terapija je bila pažljivo dizajnirana kako bi djelovala isključivo na somatske stanice, a ne na reproduktivne, čime se sprječava nasljeđivanje promjena. Unatoč obećavajućim rezultatima postignutim u ovom recentnom slučaju, postoje brojni izazovi koji se moraju riješiti prije nego što se peronalizirana terapija temeljena na CRISPR-Cas9 tehnologiji nađe u široj primjeni. Prethodni primjer proveden je na stanicama jetre koje su zahvalnije za terapiju koja se temelji na lipidnim nanočesticama u koje su bile upakiranje terapijske molekule. Razvijanje terapija koje bi učinkovito djelovale na druge dijelove tijela osim jetre su nešto na čemu će još poprilično trebati raditi.
Kako su funkcije gena na kromosomu 21 raznorodne, klinička prezentacija zahvaća sve organske sustave te je stoga jednoobrazna terapija do danas bila nemoguća
Druga lijepa vijest stigla je u lipnju a tiče se osoba s dijagnozom Downovog sindroma. Radi se o genetskom sindromu koji nastaje zbog prisutnosti dodatne kopije kromosoma 21. tzv. trisomija 21. Dodatni kromosom diže razinu ekspresije gena koji se na njemu nalaze iznad uobičajenih razina što također nije dobro za normalno funkcioniranje stanice. Ovaj sindrom nasuprot prije opisanim i nije toliko rijedak te zahvaća otprilike jedno od 700 novorođenčadi. Metode prenatalne dijagnostike su danas vrlo razvijene no stvar je izbora roditelja kakvu će odluku o svojem djetetu donijeti. Kako je prethodno rečeno, genetske poremećaje je teško liječiti na način kako uobičajeno zamišljamo liječenje primjenom neke supstance koja će eliminirati sve posljedice neke bolesti. Nažalost, kako su funkcije gena na kromosomu 21 raznorodne, i klinička prezentacija zahvaća sve organske sustave te je stoga jednoobrazna terapija, ma zapravo terapija uzroka uopće, a kamoli jednoobrazna, do danas bila nemoguća. Stoga je istraživanje Ryotaro Hashizume i suradnika sa Sveučilišta Mie u Japanu u kojem su pokazali da je moguće ukloniti suvišni kromosom iz stanica metodom CRISPR-Cas9, čime se njihovo ponašanje u laboratorijskim uvjetima približilo normalnoj funkciji, tim bitnije za osobe koje moraju živjeti s ovim sindromom. U ovom su eksperimentu dizajnirane CRISPR-vodiče (guide RNAs) tako da ciljaju isključivo nepoželjni, dodatni kromosom i eliminiraju ga iz stanice. Taj pristup u CRISPR-Cas9 tehnologiji naziva se alel-specifično uređivanje i omogućuje usmjeravanje enzima za rezanje samo na točno određenu kopiju. Tretirane matične stanice kože vratile su se uobičajenim obrascima sinteze proteina i pokazale bolju stopu preživljavanja, što upućuje na to da je genetski teret trisomije učinkovito smanjen.
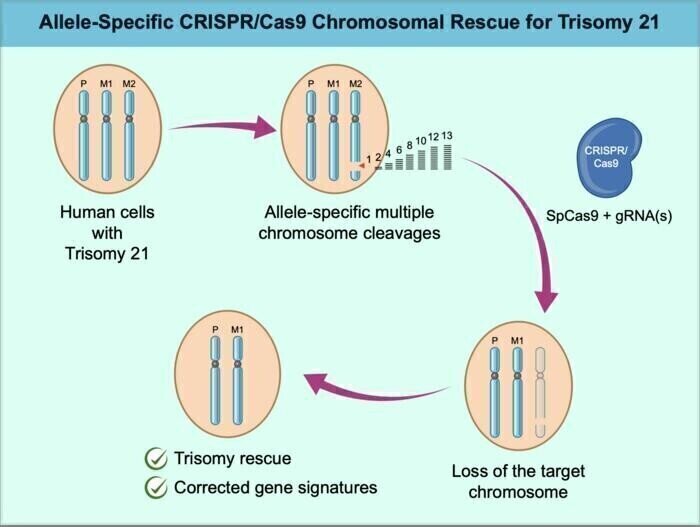
Ovaj je pristup još daleko od kliničke primjene no znanstvenici razmatraju mogućnost da bi se slična uređivanja mogla provoditi i u stanicama koje formiraju mozak i druga tkiva. Osim na matičnim stanicama pristup je ispitan i na fibroblastima kože, zrelim, diferenciranim stanicama preuzetim od osoba s Downovim sindromom te je čak i u tim potpuno razvijenim stanicama, metoda uspješno uklonila dodatni kromosom u značajnom broju slučajeva. Istraživači su također pratili kako se mijenja genska aktivnost nakon uklanjanja dodatnog kromosoma. Uočeno je da su geni povezani s razvojem živčanog sustava postali aktivniji, dok su oni vezani uz metabolizam pokazali smanjenu aktivnost. Ova promjena genske ekspresije mogla bi objasniti kako korekcija kromosomske neravnoteže utječe na cjelokupno ponašanje stanice, te potvrđuje prethodne spoznaje kako višak kromosoma 21 narušava razvoj mozga već u ranom fetalnom razvoju. Međutim, ovaj zahvat može imati i neželjne posljedice, primjerice, može zahvatiti više od jednog kromosoma 21 u stanici pa se na kraju može dogoditi i njegov manjak. Također treba spriječiti sustave za popravak DNA u stanici da ponište ili pogrešno poprave željene izmjene. Dodatni problem jest što se ovdje ne radi o pukom ispravljanju genetskih funkcionalnosti u odrasloj dobi, već mnogi od gena na ovom kromosomu imaju ključnu ulogu tijekom embrionalnog i fetalnog razvoja stoga se postavlja pitanje koristi od postnatalnih intervencija. Ipak, koristi bi moglo biti, ako već ne na mentalni razvoj, onda barem na funkcionalnost drugih tkiva, poput srčanog mišića, koji je također zahvaćen ovim sindromom.
Oba primjera u nekoliko proteklih mjeseci ove godine otvaraju mogućnosti za individualiziranu medicinu gdje terapije više ne ovise o populacijskoj učestalosti neke bolesti već o molekularnoj dijagnozi. Time daju nadu oboljelima od rijetkih i neizlječivih bolesti da će se tijekom njihovog života pronaći pristupi koji, ako ih već neće u potpunosti izliječiti, a onda će barem ublažiti simptome njihovih bolesti.
1) Musunuru K, Grandinette SA, Wang X, Hudson TR, Briseno K, Berry AM, Hacker JL, Hsu A, Silverstein RA, Hille LT, Ogul AN, Robinson-Garvin NA, Small JC, McCague S, Burke SM, Wright CM, Bick S, Indurthi V, Sharma S, Jepperson M, Vakulskas CA, Collingwood M, Keogh K, Jacobi A, Sturgeon M, Brommel C, Schmaljohn E, Kurgan G, Osborne T, Zhang H, Kinney K, Rettig G, Barbosa CJ, Semple SC, Tam YK, Lutz C, George LA, Kleinstiver BP, Liu DR, Ng K, Kassim SH, Giannikopoulos P, Alameh MG, Urnov FD, Ahrens-Nicklas RC. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. N Engl J Med. 2025 Jun 12;392(22):2235-2243. doi: 10.1056/NEJMoa2504747. Epub 2025 May 15.
2) Hashizume R, Wakita S, Sawada H, Takebayashi SI, Kitabatake Y, Miyagawa Y, Hirokawa YS, Imai H, Kurahashi H. Trisomic rescue via allele-specific multiple chromosome cleavage using CRISPR-Cas9 in trisomy 21 cells. PNAS Nexus. 2025 Feb 18;4(2):pgaf022. doi: 10.1093/pnasnexus/pgaf022.
 5.00
(2)
5.00
(2)
10. travnja 2026. / U fokusu

7. travnja 2026. / U fokusu

3. travnja 2026. / U fokusu

31. ožujka 2026. / U fokusu

10. travnja 2026. / Perspektive Rasprave

7. travnja 2026. / Perspektive Publikacije

2. travnja 2026. / Perspektive Publikacije

2. travnja 2026. / Perspektive Publikacije

{kind=link}