NOBELOVA NAGRADA ZA KEMIJU 2024.
Umjetna inteligencija riješila je najveći problem biokemije, ali – kako? To ne znamo.
 Marko Močibob / 14. listopada 2024. / Perspektive / čita se 16 minuta
Marko Močibob / 14. listopada 2024. / Perspektive / čita se 16 minuta

Marko Močibob / 14. listopada 2024. / Perspektive / čita se 16 minuta
Zahvaljujući AI-algoritmima baza riješenih struktura proteina u samo je tri godine narasla sa 100.000 na više od 200 milijuna, piše Marko Močibob. Ovogodišnji dobitnici Nobelove nagrade za kemiju riješili su jedan od najdugovječnijih izazova biokemije za sve praktične potrebe, no nismo se približili razumijevanju pravila koja upravljaju strukturama proteina. Možda ih neuronska mreža i razumije, no tu je spoznaju nemoguće izvući i prevesti na nama razumljiv način.
Polovica Nobelove nagrade iz kemije za 2024. godinu dodijeljena je Davidu Bakeru za računalni dizajn proteina, a druga polovica Demisu Hassabisu i Johnu Jumperu za predviđanje strukture proteina.
Proteini su velike biološke molekule koje u živim organizmima obavljaju najraznovrsnije poslove: hemoglobin u krvi prenosi kisik, antitijela prepoznaju ‘strance’ i pokreću imunološki odgovor koji nas brani od invazije patogenih organizama, inzulin prenosi informaciju o razini glukoze u krvi, te koordinira i podešava njen utrošak na razini cijelog organizma. Enzimi djeluju kao biokatalizatori, ubrzavaju i usmjeravaju složeni niz kemijskih reakcija koje nazivamo metabolizam. Keratin na površini kože štiti nas od vanjskih utjecaja i predstavlja granicu našeg organizma prema vanjskom svijetu. Kolagen daje elastičnost našoj koži, tkivima, ali i kostima, a miozin omogućava kontrakciju mišića i kretanje. Proteini poput transkripcijskih faktora aktiviraju i gase aktivnost pojedinih gena ili skupina gena, a drugi proteini umnažaju, popravljaju i održavaju DNA u kojoj je zapisana nasljedna informacija. Proteaze razgrađuju druge proteine kada više nisu potrebni, ili proteine iz hrane. Doista, proteini su glavni “operativci” u svim biološkim procesima i upravljaju svim aspektima onog što nazivamo život.
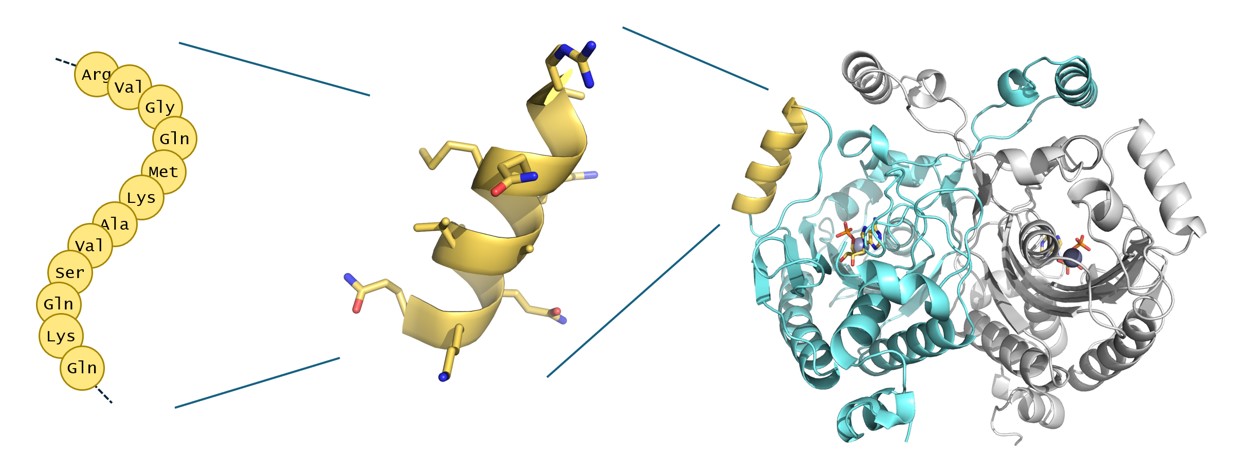
Kemijski gledano, kovalentna struktura proteina relativno je jednostavna (Slika 1): to su ravnolančani polimeri građeni od dvadeset aminokiselina. No, ti jednostavni kovalentni lanci aminokiselina se savijaju, poprimaju određene karakteristične konformacije (položaj u prostoru) i u konačnici stvaraju kompaktne, složene i jedinstvene prostorne strukture, koje se mogu dalje udruživati u još veće i složenije supramolekulske strukture. Upravo je ta trodimenzionalna, prostorna struktura proteina presudna za njihovu biološku ulogu, i prostorna struktura proteina jest ono što je evolucijski očuvano, i(li) što se tijekom evolucije dalje prilagođava.
Razumjeti kako protein funkcionira nemoguće je bez razumijevanja prostorne strukture proteina. Iako je još 1961. godine Christian B. Anfinsen pokusima pokazao da je prostorna, trodimenzionalna struktura proteina zapisana u njihovu slijedu aminokiselina (Nobelova nagrada za kemiju 1972. godine), predvidjeti kako će se protein strukturirati, odnosno kakvu će trodimenzionalnu strukturu poprimiti nije nimalo jednostavno. Za početak, taj linearni lanac aminokiselina ima nevjerojatnu konformacijsku slobodu i može poprimiti nezamislivo veliki broj različitih konformacija u prostoru. To je istaknuo američki znanstvenik Cyrus Levinthal. Izračunao je da kada bismo imali relativno mali protein od 100 aminokiselina, protein bi mogao poprimiti 1047 različitih prostornih struktura (konformacija). Kada bi protein svoju nativnu konformaciju poprimao slučajnim traženjem ili savijanjem kovalentne okosnice, bilo bi potrebno dulje od starosti svemira da pronađe ispravnu. I računalno pretraživanje svih mogućih konformacija proteina u potrazi za ispravnom strukturom nemoguće je zbog enormno velikog broja mogućih konformacija.
Anfisenovi pokusi su pokazali da je nativna struktura proteina termodinamički najpovoljnija konformacija. No, ta nativna struktura stabilizirana je brojnim i raznovrsnim interakcijama između bočnih ogranaka različitih dijelova lanca aminokiselina, gdje svaka pojedina interakcija daje maleni doprinos stabilnosti ukupne strukture. U konačnici, stabilnost prostorne strukture proteina je marginalna, jer biološki sustavi tijekom evolucije nisu optimirali proteine da budu trajni, već da se stvaraju i razgrađuju, uklanjaju, prema trenutnim potrebama stanice. Stoga, iako je načelno moguće “izračunati” nativnu strukturu proteina kao termodinamički najpovoljniju, u praksi je nemoguće predvidjeti strukturu proteina iz osnovnih fizikalnih zakona. Primarna struktura proteina (slijed aminokiselina) određuje prostornu strukturu proteina (Slika 1), ali ne postoje jednostavna “pravila” kako slijed aminokiselina prevesti u strukturu proteina, jer strukturu proteina određuju brojne i složene međusobne interakcije aminokiselina i kovalentne okosnice proteina u prostoru.

Sve to nije obeshrabrilo generacije znanstvenika da se uhvate u koštac s izazovom predviđanja strukture proteina iz njihovog slijeda aminokiselina (sekvence), iz dva razloga. Prvi razlog je fundamentalne prirode: za funkcioniranje proteina presudna je njihova prostorna struktura; ovladati sposobnošću predviđanja njihove strukture, značilo bi da doista razumijemo zakonitosti koje određuju strukturu proteina. Drugi razlog je praktične prirode: eksperimentalno određivanje prostorne strukture proteina je mukotrpan posao koji često zahtijeva mjesece ili godine posla, s neizvjesnim rezultatom.
Velik dio proteina, poput membranskih proteina ili proteinskih kompleksa, posebno je težak i nepogodan za eksperimentalno rješavanje strukture, i tek je nedavno postao dostupan strukturnoj karakterizaciji, razvojem krioelektronske mikroskopije (Nobelova nagrada za kemiju 2017. godine). K tome, eksperimentalno rješavanje strukture proteina zahtijeva posebnu infrastrukturu poput sinkrotrona, koje posjeduju samo najrazvijenije nacije, ili namjenskih, posebnih elektronskih mikroskopa koji zbog cijene nabave i održavanja često predstavljaju nacionalni resurs ili dijeljeni resurs velikih i imućnih znanstvenih ustanova (Slika 2). Stoga ne čudi da postoji velik nesrazmjer između poznatih proteina (oko 250 do 360 milijuna javno dostupnih sekvenci)[1] i broja proteina čija je struktura eksperimentalno riješena (131 000 različitih proteina, u 226 000 riješenih i javno dostupnih struktura)[2], te da postoji snažna motivacija za računalno predviđanje strukture proteina iz slijeda aminokiselina (sekvenci).
Znanstvenici su krenuli od postojećih struktura, “učeći” iz dotad riješenih struktura kako se aminokiseline međusobno “pakiraju”, kakvi se strukturni motivi u proteinima pojavljuju, minimizirajući energiju mogućih konformacija, koristeći sve dostupne trikove i prečice kojima su se mogli domisliti (podsjećam da je “izračunati” ispravnu strukturu teorijski moguće, ali u praksi neostvarivo). I tako su kroz desetljeća polako napredovali, i postali prilično dobri u predviđanju struktura proteina, ako je u bazama podataka riješenih struktura (Protein Data Bank) već prisutan neki sličan protein. Naime, često je prostorna struktura evolucijski očuvana čak i ako se sličnost na razini sekvence proteina izgubila. Na tome se temelji tzv. homologijsko modeliranje. Nadalje, neki strukturni motivi se ponavljaju u različitim proteinima. No, predvidjeti strukturu istinski novog proteina, različitog od dotad riješenih struktura, ili pronaći odgovarajući predložak u Protein Data Bank ostalo je nedostižno, i tu su različiti algoritmi i pristupi redovito zakazivali.
Velika pomoć i nezamjenjiv alat bila su naravno računala, radi se o računalnim programima, naprednim i zahtjevnim informatičkim algoritmima. Godine 1994. započela je organizirana usporedba i vrednovanje različitih pristupa i algoritama različitih istraživačkih grupa, pod nazivom Critical Assessment of Protein Structure Prediction (CASP), koja je prerasla u svojevrsno natjecanje, svake dvije godine.
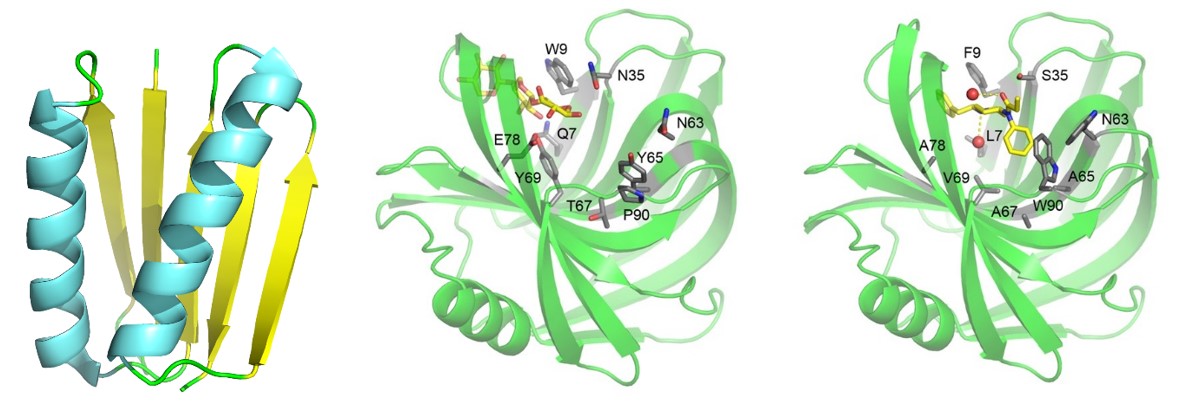
No problemu razumijevanja strukturiranja proteina moglo se pristupiti i obrnuto, u obliku tzv. The Inverse Protein Folding Problem: ako znamo kakav protein želimo, možemo li predvidjeti slijed aminokiselina koji će dati takvu strukturu? Kako se slikovito izrazio David Baker, jedan od ovogodišnjih laureata: “If you want to build an airplane, you don’t start by modifying a bird; instead, you understand the first principles of aerodynamics and build flying machines from those principles.“[3] (Ako želiš napraviti avion, ne krećeš od preinake ptica, već nastojiš shvatiti zakone aerodinamike i na temelju njih konstruirati leteće strojeve.) Analogija je izvrsna, jer su se ljudi otisnuli od zemlje u zrak kad su prestali mahati krilima. Davidu Bakeru i suradnicima to je pošlo za rukom 2003. godine (Slika 3). David Baker veteran je računalne strukturne biologije i dugogodišnji sudionik CASP-a, na kojem je njegova Rosetta postizala zapažene rezultate. On je odlučio svoj algoritam upotrijebiti u obrnutom smjeru: za zadanu strukturu, koja dotad nije postojala u bazama podataka, naći slijed aminokiselina koji će poprimiti takvu prostornu strukturu. Kada je kandidat proizveden u živom sustavu, lanac aminokiselina doista je posjedovao zamišljenu strukturu.
Kako su to postigli? Njegov program Rosetta u postojećim nevezanim proteinima tražio je male strukturne fragmente slične sekvence, te ih paralelno optimirao na razini sekvence i strukture prema konformaciji ciljne strukture. Konformacija bočnih ogranaka uzorkovana je iz velike biblioteke rotamera, te su Monte Carlo simulacijama optimirani suptilni učinci dodirivanja hidrofobnih aminokiselina, vodikovih veza i utjecaja otapala. S vremenom, David Baker i suradnici producirali su niz potpuno novih, dotad nepoznatih proteina željene strukture, ali i sposobnosti da npr. vežu male molekule poput danas zloglasnog fentanila (jak analgetik, ali i ilegalna droga; Slika 3). Godine 2008. čak su uspjeli dizajnirati enzim, doduše relativno spor u usporedbi s prirodnim enzimima, ali koji je katalizirao kemijsku reakciju za koju prirodni enzimi nisu postojali.
No, vratimo se predviđanju strukture proteina: u prirodi postoji toliko velik broj različitih proteina da većinu istraživača ipak više zanima predvidjeti strukturu proteina od interesa, ili dobiti uvid u raznolikost postojećih proteina (koji doista jesu nevjerojatno raznoliki), nego dizajnirati vlastite proteine. Napredak na tom polju bio je postojan, ali spor i inkrementalan. Prve natruhe onog što dolazi dogodile su se na CASP 2018. godine, kad se na natjecanje kao autsajder prijavio DeepMind, tvrtka čiji je osnivač Demis Hassabis, a u vlasništvu tehnološkog giganta Google. Tvrtka DeepMind razvijala je algoritme i primjene umjetne inteligencije. Prvi veliki i svjetski zapažen uspjeh tvrtke je razvoj programa AlphaGo, koji je 2016. godine pobijedio svjetskog prvaka u igri Go.
Za igru Go je karakteristično da ima neizmjerno puno mogućih kombinacija koje se brzo granaju, i da je nemoguće dobiti je sirovom računalnom snagom u predviđanju idućih koraka, jer broj mogućih koraka i scenarija vrlo brzo eskalira. Vrlo brzo su razvili i program AlphaZero (2017.), koji u nekoliko dana može savladati pravila i postati nepobjediv u praktički bilo kojoj igri tipa Go, šaha ili sl. igara na ploči. DeepMind se 2018. godine prijavio na CASP13 s programom AlphaFold i – pomeo konkurenciju u predviđanju strukture proteina. No pravi šok uslijedio je 2020. godine. Iako je prvotna verzija AlphaFolda bila najbolja platforma za predviđanje strukture proteina, DeepMind je radikalno redizajnirao svoj algoritam, počevši iz početka: korišten je drugačiji tip neuronskih mreža, drugačija računalna reprezentacija proteina tijekom rada programa, algoritam je promijenjen. Tu je značajan doprinos dao John M. Jumper, treći ovogodišnji laureat.
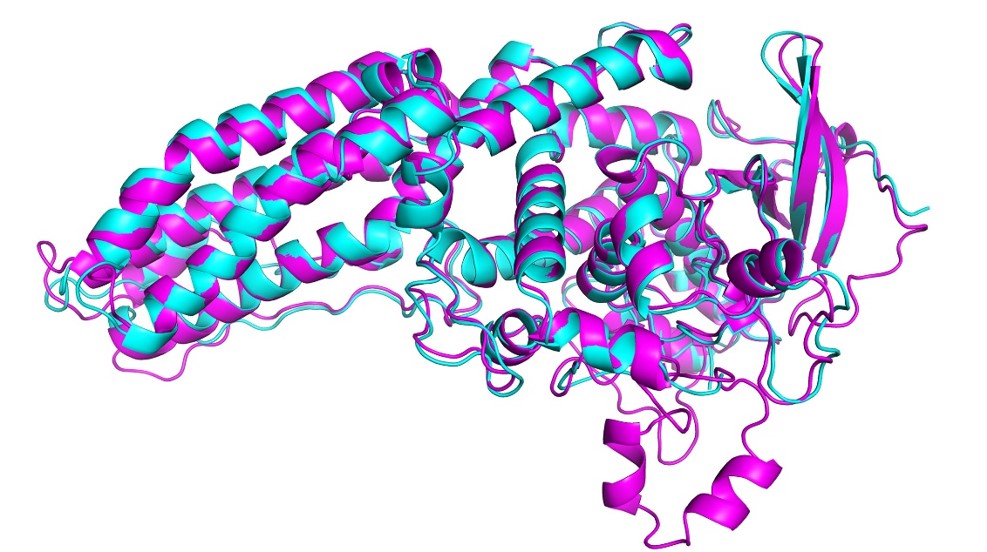
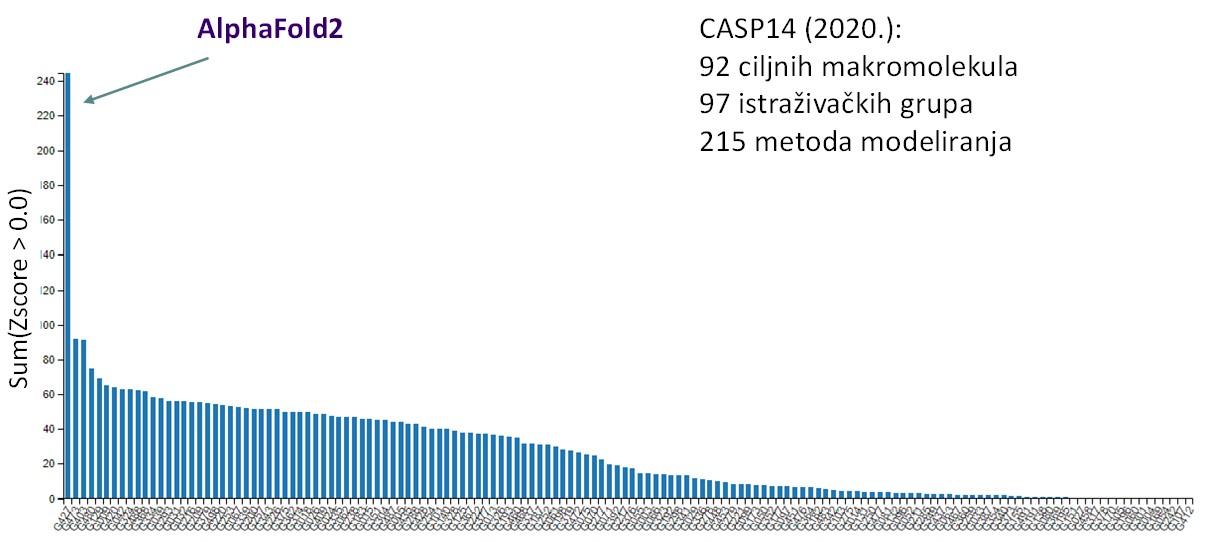
Novi program AlphaFold2 na CASP14 ne samo da je ponovno potukao konkurenciju (Slika 4), nego je daleko nadmašio i prvotni AlphaFold s CASP13, iz 2018. godine. Predviđanja struktura koje je AlphaFold2 generirao često su bile neraspoznatljive od eksperimentalno dobivenih struktura (Slika 5)! Bez da ulazimo u detalje, eksperimentalne strukture nisu “savršene”, one su isto svojevrstan model i posjeduju određenu grešku. Predviđanja koja je dao AlphaFold2 bila su toliko dobra da je često bilo nemoguće reći radi li se o stvarnim odstupanjima od stvarnih struktura, ili su odstupanja unutar neodređenosti samih eksperimentalnih struktura.
Idućih godina područje strukturne bioinformatike doživjelo je pravu revoluciju i napredak o kojem nismo mogli ni sanjati. DeepMind je sklopio partnerstvo s EBI (European Bioinformatics Institute), uglednom akademskom ustanovom, i svoje modele učinio javno dostupnima: sredinom 2021. objavili su sveukupno 350.000 struktura proteina iz čovjeka i 20 najvažnijih modelnih organizama u biologiji, u prosincu 2021. modelirali su cijeli Swiss-Prot, važnu bazu podataka proteinskih sekvenci, a broj predikcija struktura porastao je na 800.000. U srpnju 2022. objavili su ukupno 214 mil. predikcija, gotovo cijeli UniProt 2021_04, što praktički predstavlja sve poznate proteine. Nadalje, javno su objavili kako AlphaFold radi, pa su i ostali, npr. David Baker, svojevrsnim reverznim inženjeringom razvili slične programe (RoseTTAFold, RoseTTAFold2), koji su postizali jednake, ili gotovo jednako spektakularne rezultate.

Sredinom 2021. godine učinili su i računalni kod javno dostupnim, pa su se pojavili i slobodno dostupni serveri i unaprijeđene inačice AlphaFolda2 koje su bile brže od izvorne, ili su mogle modelirati i multimerne proteine (npr. ColabFold). Znanstvena zajednica je nevjerojatno brzo prigrlila novi alat, i njegov disruptivni potencijal je odmah prepoznat te je časopis Science AlphaFold2 i slične programe, odnosno primjenu umjetne inteligencije u predviđanju strukture proteina proglasio za Breakthrough of the Year 2021 (Slika 6).U vrlo kratkom vremenu, od svega 2-3 godine (od 2020. do 2022.), istraživačima je od cca. 100.000 eksperimentalno riješenih struktura 2020. godine postalo dostupno preko 200 milijuna struktura, kvalitetom usporedivih s eksperimentalnima, praktički bilo kojeg poznatog proteina.
Ali kako je AlphaFold2 postigao takav proboj u području predviđanja strukture proteina? Paradoksalno, ni u jednom svom aspektu AlphaFold2 nije revolucionaran; što ne umanjuje značaj tog alata, napor i inventivnost njegovih tvoraca. Riječima samog Johna M. Jumpera, jednog od tvoraca AlphaFold2: “It wasn’t just that we went to work and we pressed the AI button, and then we all went home. It was really an iterative process where we developed, we did research, we tried to find the right kind of combinations between what the community understood about proteins and how do we build those intuitions into our architecture.“[4] (Nismo došli na posao i samo stisli dugme za AI, pa svi otišli kući. Bio je to iterativan razvoj, istraživali smo, pokušavali naći pravu kombinaciju onoga što [znanstvena] zajednica razumije o proteinima, i kako te spoznaje ugraditi u našu [računalnu; softversku] arhitekturu.)
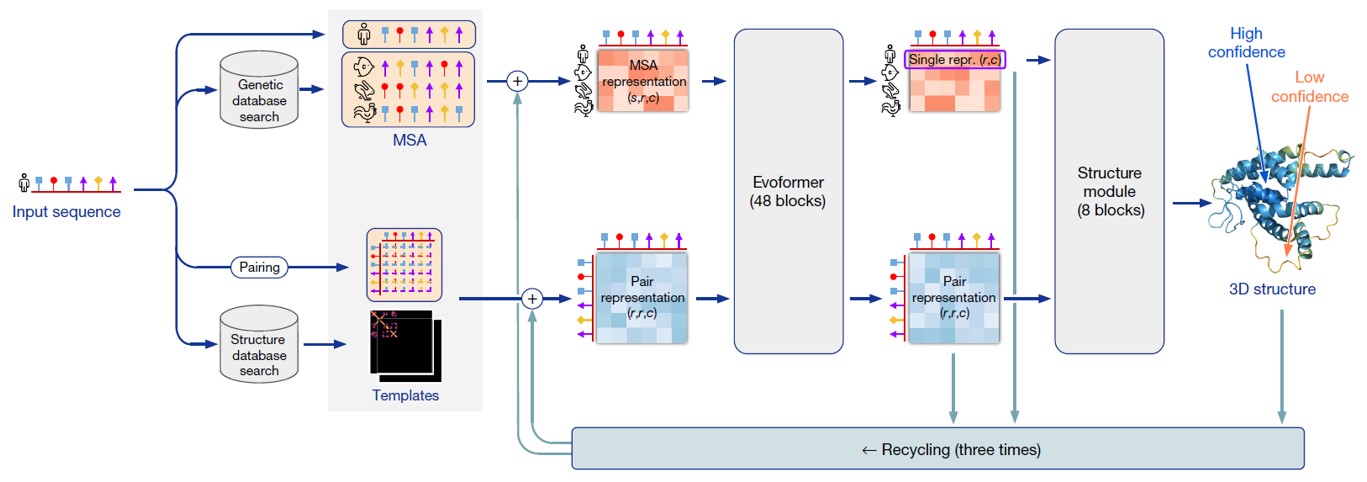
Njegovi tvorci, Demis Hassabis, John M. Jumper i suradnici primijenili su u razvoju Alphafold2 moderne i sofisticirane oblike strojnog učenja temeljenog na transformerima, jedne vrste neuronskih mreža (slika 7). Nadalje, uspješno su iskoristili otprije poznat princip kovarijacije, činjenicu da su mutacije aminokiselina na prostorno bliskim položajima u proteinu međuovisne, i “zapisane” u evolucijskim trajektorijama proteina, te veliku količinu javno dostupnih podataka o strukturama proteina pohranjenih u Protein Data Bank za treniranje svojeg modela. Za treniranje, odnosno ugađanje takvih dubokih modela strojnog učenja potrebne su velike količine podataka, tj. riješenih struktura, i očito je dostignuta ta kritična količina podataka da razvoj takvog sustava dubokog strojnog učenja ili umjetne inteligencije postane moguć. Također, nedvojbeno su ključni bili i praktično neograničeni računalni resursi kojima DeepMind raspolaže. Ali, ništa od navedenog nije zamjena niti može kompenzirati ljudski genij, odnosno dvojac koji je prepoznao trenutak, te posjedovao ekspertizu i ambiciju da to ostvari.
Od Anfisenovih pokusa i prvih eksperimentalno riješenih struktura proteina krajem 50-tih i početkom 60-tih godina 20. stoljeća prošlo je oko 65 godina. Jesmo li konačno riješili jedan od najvećih i najdugovječnijih izazova u biokemiji, znati predvidjeti strukturu proteina? Odgovor na to pitanje uvelike ovisi kako točno pitanje formuliramo. Za sve praktične potrebe – DA!
Sustavi umjetne inteligencije temeljeni na strojnom učenju poput AlphaFold2, RoseTTAFold2, ESMFold i drugi trenutno produciraju modele koji su iznimno kvalitetni, a razvijeni su i sustavi koji daju modele kompleksa proteina, kompleksa s nukleinskim kiselinama, malim molekulama, itd. Ti modeli su jako korisni u radu istraživača i mogu u brojnim situacijama zamijeniti eksperimentalno određene strukture.
No jesmo li doista i istinski naučili kako predvidjeti strukturu proteina?
Zapravo, NE: priroda neuronskih mreža i sustava dubokog strojnog učenja takva je da iz njih ne možemo izvući, apstrahirati, “pravila” koja su u tu mrežu urezana treningom, niti prevesti na nama razumljiv i shvatljiv način. Te poveznice između čvorova neuronskih mreža, način kako rade, k tome je svojstven i vezan za konkretnu neuronsku mrežu ili sustav dubokog strojnog učenja. Ne mogu se jednostavno prenijeti ili presnimiti u novi, napredniji sustav, već se nova mreža, sustav ili algoritam opet treba istrenirati iznova, iznova testirati. Koliko god AlphaFold2 i srodni sustavi dobro radili, ostaje neugodan osjećaj da se njima koristimo kao crnom kutijom. Postoje mišljenja da AlphaFold2 funkcionira na višoj razini od prepoznavanja obrazaca (pattern recognition) i da je u svoje neuronske mreže doista uhvatio fizikalne principe koji ravnaju proteinskim strukturama, no to znanje (ako postoji) nećemo moći dobiti ili izdvojiti iz sustava umjetne inteligencije.
I nekoliko zanimljivosti za kraj: u svibnju 2024. godine izašla je nova verzija AlphaFold3, i prateći članak u časopisu Nature. Veliko negodovanje akademske zajednice izazvalo je to što izvorni kod AlphaFold3 nije objavljen, a nije bio dostupan čak ni recenzentima tijekom recenzije. Dakle, nitko ne zna kako točno novi AlphaFold radi, niti je mogao provjeriti navode autora članka. Umjesto slobodnog koda, DeepMind je ovaj put odlučio omogućiti korištenje njihova sustava, ali uz određena dnevna i druga ograničenja. Najavljeno je da će “AlphaFold3 model” biti objavljen unutar 6 mjeseci za akademsku upotrebu, ali u trenutku pisanja ovog članka to se još nije desilo. Najvažnija nova odlika AlphaFold3 naprema verzije 2 jest da ima mogućnost modeliranja biomolekulskih interakcija sa svim vrstama liganada, malih molekula i modifikacija proteina, tj. da ima mogućnost modeliranja – predviđanja interakcije proteina i farmaceutski aktivnih tvari, s neslućenim potencijalom za razvoj lijekova. DeepMind je odlučio komercijalizirati takvu primjenu kroz partnerstvo s tvrtkom za razvoj lijekova Isomorphic Labs, u vlasništvu Alphabeta, krovne tvrtke Googlea.
Umjetna inteligencija preporodila je i područje de novo dizajna proteina, u režiji Davida Bakera, čija je istraživačka grupa razvila RoseTTAFold diffusion (RFdiffusion), generativni AI koji funkcionira na sličan način kao Midjourney, DALL-E, Stable Diffusion ili slični generativni sustavi umjetne inteligencije koji na zadani upit daju fotorealistične ili imaginarne slike. Rezultati su zbilja impresivni, kada su takvi proteini proizvedeni u laboratoriju potvrđeno je da imaju očekivanu strukturu. Zanimljivo, RFdiffusion i primjena generativne umjetne inteligencije u dizajnu proteina nije obuhvaćena dodijeljenom Nobelovom nagradom.
https://www.nobelprize.org/prizes/chemistry/2024/popular-information/
https://www.nobelprize.org/uploads/2024/10/advanced-chemistryprize2024.pdf
https://www.nature.com/articles/s41580-019-0163-x
[1] Podaci iz baze podataka UniProt, za release 2024_05, i NCBI Reference Sequence Database, na dan 15. listopada 2024. Koliko (različitih) proteina je danas poznato nije trivijalno pitanje, jer različiti organizmi mogu posjedovati identične proteine. Različite baze podataka različitim strategijama pristupaju tom pitanju i problemu redudancije podataka, i što uvrštavaju u svoju bazu, pa se i konkretni brojevi razlikuju.
[2] Broj struktura različitih proteina, ili broj jedinstvenih struktura je manji od ukupnog broja eksperimentalno riješenih struktura pohranjenih u PDB, jer pojedini protein može biti kristaliziran ili njegova struktura riješena u kompleksu s različitim drugim molekulama, na različitim rezolucijama, različitih mutanata i sl. Iako takve strukture imaju svoju svrhu i vrijednost, u ovom kontekstu one ne predstavljaju istinske nove ili različite strukture.
[3] https://www.nobelprize.org/prizes/chemistry/2024/popular-information/
 5.00
(2)
5.00
(2)
31. ožujka 2026. / U fokusu

25. ožujka 2026. / U fokusu

24. ožujka 2026. / U fokusu

17. ožujka 2026. / U fokusu

28. ožujka 2026. / Perspektive

27. ožujka 2026. / Perspektive Video

27. ožujka 2026. / Perspektive Publikacije

23. ožujka 2026. / Klub Batina Perspektive Rasprave



{kind=link}